





| A 45 year-old Woman with an Enhancing Brain
Mass. June, 2003, Case 306-1. Home Page |
Walter F. Bierbaum, M.D.1, Kar-Ming Fung, M.D., Ph.D.1, Gregory N. Fuller, M.D., Ph.D.2 Last update: June 30, 2003.
1 Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma, 2 Department of Pathology, University of Texas M.D. Anderson Cancer Center, Houston, Texas
Clinical information: The patient was a 45 year old woman with a headache. MRI revealed a strongly enhancing dural based tumor of the falx cerebri. Edema was noted in the surrounding brain tissue but no mass effect was noted. Surgery yielded the following specimen.
Histopathology of the case:
|
|
|
|
|
|
|
| A. | B. | C. | D. | E. | F. |
|
|
|
|
|||
| G. | H. | I. |
Panel A and B are low-magnification photographs taken at an area adjacent to the dura. Panel C, D, and E are taken from similar areas. Panel F, G, H, and I are taken from similar areas.
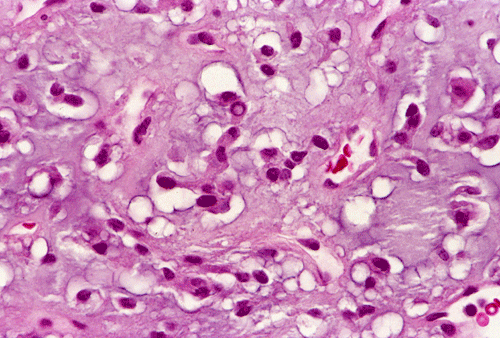
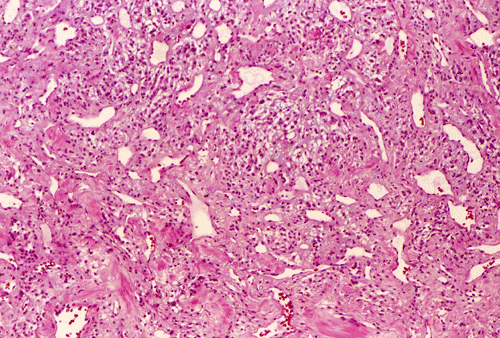
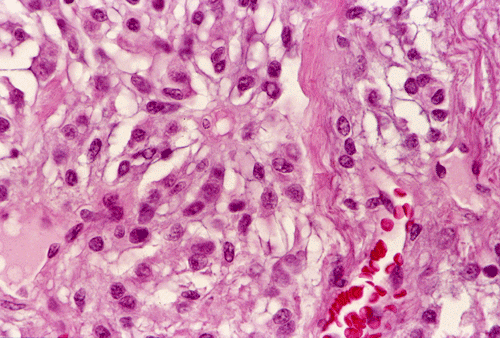
The tumor was removed as multiple tissue fragments. At low-magnification, the lesion appeared as a cellular neoplasm (Panel A) with involvement of the dura (Panel B). There was a variation of histopathologic features in different areas of the specimen. In a number of areas, the histopathologic features were similar to those in Panel C, D, and E. In essence, the lesion was a cellular neoplasm with a pale blue myxomatous background. The tumor cells varied from round/polygonal to spindle-shaped (Panel D). The amount of pale blue myxomatous background also varied (compare Panel C and D). The tumor had small hyperchromatic and bland nuclei without prominent nucleoli. In some tumor cells, the cytoplasm contained several small vacuoles that gave them a bubbly appearance. In other tumor cells, the cytoplasm was dominated by one or two large vacuoles (Panel E).
In other areas (Panel F, G, H, and I), the tumor had increased vascularity with sinusoidal vascular spaces. No anastomosing vascular channels were found (Panel F, G, and H). The volume of myxomatous background substance was reduced. The tumor cells appeared epithelioid and contained bubbly to clear cytoplasm. The nuclei had coarse to clumpy chromatin. Occasional intranuclear pseudoinclusions could be found (Panel H, and I).
Neither necrosis nor endothelial proliferation was noted in any part of the tumor. Mitotic figures were not readily seen. Very little brain parenchymal tissue was adherent to the tumor and brain invasion could not be evaluated.
| DIAGNOSIS: Chordoid meningioma (WHO grade II/IV). | i How to approach this case? |
Discussion: General Information Meningioma in general Chordoid meningioma Differential diagnosis
General Information
Meningiomas are neoplasms that display phenotypic features of arachnoid (meningotheliatous) cells. Most meningiomas are intracranial but about one-tenth of them occur in the spinal cord, very few of them arise from the ventricles. Some rare examples are found outside the central nervous system such as the lung, the mandible, and the scalp. Meningiomas are very common and comprise about one-fifth of all primary brain tumors. About one-tenth of meningiomas are multiple at the time of presentation. The peak incidence is between 45 to 55 years of age and there is a clear female predilection. Although meningiomas are seen in all age groups, they are uncommon in children and almost unknown in infants. When they occur in the very young, however, they tend to behave more aggressively. In the 4 tier system of the World Health Organization (WHO) Classification of Tumors, three major histologic grades (WHO grade I, II, and III) for meningiomas with increase in anaplastic features and aggressive biological behavior from grade I to III. Recurrence of a WHO grade I meningioma after surgery is not uncommon. Meningiomas are seen in about half of the patients with neurofibromatosis 2 (NF2) but are not associated with neurofibromatosis 1 (NF1).
The
intracranial distribution of meniningiomas corresponds with the
distribution of the arachnoidal granulations. Most of them are found firmly
affixed to the dura with half of all cases adjoining the sagittal sinus. The
rest are predominantly seen in other convexities of the brain. Other common
locations include olfactory grooves, the tubeculum sellae and parasellar region,
the dural sheath of the optic nerve and the petrous bone. Meningiomas may also
arise in the tela choroidae or in the stroma of the choroids plexus as
intraventricular tumors. Spinal meningiomas are usually laterally situated,
intradural, extramedullary tumors with a close relationship to the nerve roots.
Macroscopically and radiographically, spinal meningiomas often raise the
possibility of schwannoma.
In
computerized axial tomograph (CAT) scans, about three-quarter of meningiomas are
hyperdense with calcifications found in about one-quarter of all cases. While
they appear isointense with gray matter in T1-weighted non-contrast enhanced
magnetic resonance imaging (MRI), they enhance brightly, and often homogenously.
About slightly more than half of the cases have enhancement in the adjacent dura
(the so-called “dural-tail”). A crest of cerebral spinal fluid (CSF)
separating the tumor from the brain parenchyma (the so-called “CSF crest”)
is often, but not always, present and is best detected in T2-weighed images.
Edema in the brain parenchyma surrounding the tumor is quite variable. Invasion
into the surrounding cranial bone is not an uncommon feature and is not an
indication of biological malignancy. These cases, however, are more surgically
challenging.
Pathology of meningiomas in general
Embryologically,
the meninges are condensations of the mesenchymal elements around the neural
tube. The telencephalic leptominges and the dermis of the upper face arise from
the neural crest cells. The leptominges of the posterior brain and the spinal
cord arise from the mesoderm. The role of the arachnoid cell in the
cerebrospinal fluid circulation and its function as part of the brain coverings
require both epithelial and mesenchymal properties.
This is clearly reflected in the morphologic features of arachnoid cells
particularly at the ultrustructural level. As
a family, there are probably no other tumors in the central nervous system that
display a richer diversity in histopathologic features. The biological behavior
can be correlated with some histologic variants. While the typical meningiomas
are easy to diagnose, the uncommon variants often mimic other tumors and pose
diagnostic difficulties. The
rich diversity in histopathologic features may well be related to the diverse
embryonic and biological features of arachnoid cells.
Most
meningioms display more than one histologic pattern and rarely exist in one pure
histologic pattern. The most common histologic types are the meningothelial type
and mixed type. The fibrous (fibroblastic) type is less common. The
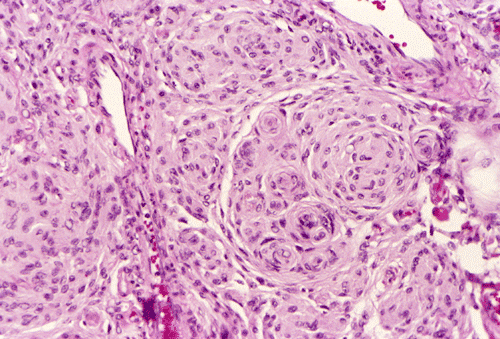
meningothelial type has morphologic features similar to arachnoid cells.
Typically, they are composed of bland syncytial epithelioid cells. The nuclei
are oval, with distinct nuclear membrane, and fine chromatin. A single nuclear
groove is seen sometimes. Pseudonuclear inclusions resulting from protrusion of
cytoplasm into the nuclei are often present. Nucleoli are indistinct, if
present. The neoplastic meningothelial cells tend to arrange in concentric
whorls. The fibroblastic pattern has long spindle cells and fibroma like
morphology. These features are illustrated in the box below. Ill-defined palisades of tumor cells suggestive of schwannoma may
occur. With careful search, meningotheliomatous whorls that help confirm the
diagnosis of meningioma are often found in fibrous meningiomas. The
mixed type bears features of the meningothelial type and
fibroblastic type. Psammoma bodies are often found at the center of the
meningothelial whorls. When they are abundant, the term psammomatous meningioma
is used. The psammoma bodies may become confluent and even ossified. Mesenchymal
metaplastic changes including as osseous, cartilaginous, lipomatous, myxomatous
and xanthoumatous changes can occur and complicate the otherwise classic
meningiomas.
 |
 |
 |
 |
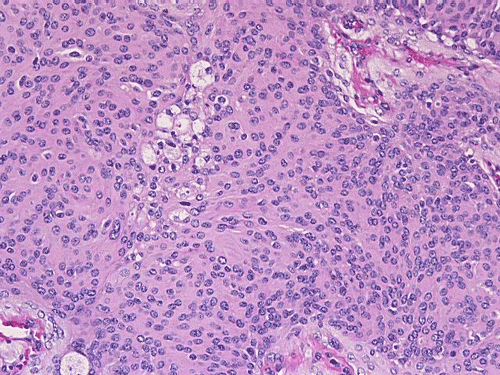
| Click thumbnail to see meningothelial whorls in a meningioma. | Click thumbnail to see meningiothelial whorls in high-magnification. Note the nuclear pseudoinclusions (arrow). | Click thumbnail to see a meningothelial meningioma with epithelioid pattern ill-defined meningothelial whorls. | Click thumbnail to see a fibrous meningioma. |
Several
other distinct morphologic variants of meningiomas are also recognized. While
the angiomatous meningioma, microcystic meningioma, secretory meningioma, and
lymphoplasmacyte-rich meningioma have distinctly different morphologic features,
they have biologic behavior similar to that of the meningothelial, mixed, and
fibrous meningioma and are WHO grade I tumors. In contrast, chordoid meningioma
and clear cell meningioma (particularly those associated with an intracranial
location) behave less favorably and are recognized as WHO grade II tumors.
Papillary meningioma and rhabdoid meningiomas tend to behave aggressively and
are recognized as WHO grade III tumors.
Although
some pleomorphism and mitotic activities may be noted in any of these variants,
these features do not necessarily connote aggressive behavior. The WHO
classification also recognizes atypical meningioma (WHO grade II) and anaplastic
(malignant) meningioma (WHO grade III). The diagnostic criteria of atypical and anaplastic meningioma
are independent of the meningioma subtype. The WHO classification defined atypical
meningioma as “A meningioma with increased mitotic activity or three or more
of the following features: increased cellularity, small cells with high
nucleus:cytoplasm ratio, prominent nucleoli, uninterrupted patternless or
sheet-like growth, and foci of spontaneous or geographic necrosis.” Anaplastic
meningioma is defined as “A meningioma exhibiting histological features of
frank malignancy far in excess of the abnormalities present in atypical
meningioma.” In our opinioin, a gray zone exists in between meningioma and
atypical meningiomas.
Pathology of chordoid meningioma
In
our case under discussion, the pale blue myxoid background and the cytoplasmic
vacuoles are features reminiscent of chordoma. This tumor was also
immunoreactive for epithelial membrane antigen (EMA). With its dural-based
origin in the convexity, the best diagnosis is chrodoid meningioma (WHO grade
II). The rich vascular feature in some parts of the tumor is consistent with and
angiomatous meningioma. The cytologic features in the vascular area, however,
are very similar to that of the chordoid component. This area is best
interpreted as chordoid meningioma with angiomatous changes rather than an
angiomatous meningioma.
Chordoid
meningioma was first described by Kepes et al. in 1988
1.
In this study, all seven patients were under 20 years of age. The tumors were
associated with dense lymphoplasmacytic cell infiltration with lymphoid
follicles and germinal center. These patients also had preoperative
manifestation of Castleman’s disease that included iron resistant microcytic
anemia, stunted growth, plasmacytosis, dysgammagloulinema, and
hepatosplenomegaly. The hematologic abnormalities of these patients normalized
as the meningiomas were removed. Subsequent studies did not confirm the
association with Castleman’s disease or other systemic manifestaion nor its
preferred occurrence in young patients. In the same study, the patient ranged
from 12 to 77 years of age and there was only minimal female predilection. The
lymphoplasmacytic infiltration was not seen in 40.5% of the cases; it ranged
from mild to moderate in the rest of the cases
2.
Sporadic reports of chordoid menigniomas with and without association with
systemic symptoms of Castleman’s disease and occurring in both adult and
children have been reported
3,4,5,6.
Gross examination of a chordoid meningioma would reveal a gelatinous consistency with
variable hemorrhage. Areas with
more features of typical meningiomas, areas with tan to gray with a whirled to
granular cut surface, may intersperse with the
gelatinous tissue. Microscopically,
chordoid meningiomas typically contain spindle to epitheliod cells arranged in
cords or sheets resembling chordoma. The
neoplastic cells often have a physalliforous (bubbly or vacuolated cytoplasm)
appearance and appear to float within a myxoid stroma.
It is uncommon for chordoid meningioma to exist as a pure form. These
chordoma-like areas usually coexist with areas with typical histologic featues
of meningiomas including syncytial meningothelial cells in whorls, psammoma bodies,
and fibrous menigothelial areas. Lymphoplasmacytic infiltration may or may not
be present. Electron microscopic
findings including slender, interdigitating cytoplasmic processes and desmosomes,
support a diagnosis of meningioma
7.
Immunohistochemically,
there is no single phenotypic marker that is specific for meningiomas. These
markers, however, are very useful in separating meningiomas from other tumors.
Most meningiomas have immunoreactivity for epithelial antigen (EMA)
8,9,10.
This is particularly true in areas with meningothelial features.
The immunoreactivity for cytokeratin has been reported in 50% of
meningiomas
8,9,10.
In our experience, the immunoreactivity is usually focal and sporadic. This is a
useful feature that separates meningiomas from metastatic carcinoma where
diffuse and strong immunoreactivity for cytokeratin is almost the rule. Only
weak and focal immunoreactivity for S-100 protein is seen in most meningiomas
other than fibrous meningioma
8
which tend to have more impressive expression of S-100 protein. Meningothelial
meningiomas tend to have immunoreactivity for many of the aforementioned
antigens. Slightly more than half of all meningiomas express progesterone
receptor. Immunodetection for estrogen receptor is usually negative. In contrast
to schwannoma, meningiomas tend to have reticulin deposition around the cell
nest but not around individual cells. A recently discovered unbalanced
translocation t(1;3)(p12-13;q11) has been found in three chordoid meningiomas
and may serve as potential diagnostic aid in future
11.
Differential diagnosis
The
differential diagnoses for chordoid meningioma include schwannoma with
myxomatous change, chordoma, and
cartilaginous tumor. Schwannomas and meningiomas are both commonly found
as intradural-extramedullary tumors. Intracranial schwannomas almost never occur
as dural-based tumor in the convexity. Schwannomas typically have reticulin
stain around individual cells and are strongly positive for S-100 protein. At
the ultrastructural level, schwannomas have electron dense basal lamina and
long-spacing collagen.
Chordomas
often arise in proximity to bony midline structures, notably the clivus and
either end of the spine. Histologically,
they are composed of the pathognomonic physaliphorous cells but some cells with
more solid and eosinophilic cytoplasm are often present. The cells are embedded
in an abundant, basophilic, metachromatic myxomatous matrix. They are strongly
immunoreactive for S-100 protein, cytokeratin, and also EMA. Chodoid chondroma
is a concept and also an entity that is of much dispute
12.
Chrondrosarcomas
arising from the cranial base are uncommon cartilaginous tumors that invade the
adjacent structures but rarely metastasize. They have a wide age range and with
slight female predilection. Half of them are found in the tempero-occipital
junction. The less common site is the clivus followed by sphenoethmoid complex. The most common
histologic type is a mixed hyaline and myxoid type. The histologic grade is
always low. Immunohistochemistry allows separation of chordoid meningioma from
chondrosarcomas. Over 95% of cranial base chondrosarcoma are positive for S-100
protein but only a few of them are immunoreactive for EMA. They are negative for
cytokeratin. These features also allow separation from chordoma.
Last but not least, ecchondrosis
physaliphora may mimic chordoid meningioma or chordoma when presented
without adequate clinical information. Ecchondrosis occurs as single ectopic
gelatinous nodule of notochordal tissue overlying the ventral aspect of the pons
in the midline. They are found in about 2% of autopsies as an incidental
finding. It has a subdural location and is attached to the basioccipital bone by
means of a fragile stalk that passes through a pore in the dura. They are poorly
nourished and often show advanced degeneration microscopically. Most chordomas
do not arise from these remnant structures.
Reference:
Kepes
JJ, Chen WY, Connors MH, Vogel FS.
"Chordoid" meningeal tumors in young individuals with peritumoral
lymphoplasmacellular infiltrates causing systemic manifestations of the
Castleman syndrome. A report of seven cases. Cancer. 1988
15;62:391-406.
Couce
ME, Aker FV, Scheithauer BW.
Chordoid meningioma: a clinicopathologic study of 42 cases. Am
J Surg Pathol. 2000 24:899-905.
Kobata
H, Kondo A, Iwasaki K, Kusaka H, Ito H, Sawada S. Chordoid
meningioma in a child. Case report. J Neurosurg. 1998 88:319-23..
Mori
S, Oka K, Hakozaki H, Soga Y, Hayano M, Oka T, Nakazato Y, Mori N. Chordoid
meningioma. A case report. Pathol Res Pract. 2001;197:515-8
de Tella OI Jr, Herculano MA, Prandini MN, Stavile JN, Bonatelli Ade P. Chordoid meningioma: report of two cases. Arq Neuropsiquiatr. 2003 61:91-4.
Yano
H, Shinoda J, Hara A, Shimokawa K, Sakai N.
Chordoid meningioma. Brain Tumor Pathol. 200017:153-7.
Zuppan
CW, Liwnicz BH, Weeks DA. Meningioma
with chordoid features. Ultrastruct Pathol. 1994 18:29-32.
Artlich A, Schmidt D. Immunohistochemical profile of meningiomas and their histological subtypes. Hum Pathol. 1990 21:843-9.
Perry
A, Scheithauer BW, Nascimento AG. The
immunophenotypic spectrum of meningeal hemangiopericytoma: a comparison with
fibrous meningioma and solitary fibrous tumor of meninges. Am J Surg
Pathol. 1997 21:1354-60.
Ng HK, Wong AT. Expression of epithelial and extracellular matrix protein markers in meningiomas. Histopathology. 1993 22:113-25.
Steilen-Gimbel
H, Niedermayer I, Feiden W, Freiler A, Steudel WI, Zang KD, Henn W. Unbalanced
translocation t(1;3)(p12-13;q11) in meningiomas as the unique feature of
chordoid differentiation. Genes Chromosomes Cancer. 1999 26:270-2.
Brooks JJ, LiVolsi VA, Trojanowski JQ. Does chondroid chordoma exist? Acta Neuropathol (Berl). 1987 72:229-35.