 Click thumbnails
to see pictures.
Click thumbnails
to see pictures. | An Infant at 30 Weeks of
Gestation and with a Rapidly Enlarging Head. July, 2005, Case 507-2. Home Page |
Jianyi Li, M.D., Ph.D, Kar-Ming Fung, M.D., Ph.D. Last update: May 30, 2005.
Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Clinical information: The mother was a 22 year-old woman. Rapid enlargement of head circumference of the fetus was noted in prenatal care. A female infant was delivered by Cesarean section after 30 weeks of gestation. The infant died several hours after delivery. Autopsy was performed and was limited to the brain.
Click thumbnails
to see pictures.
Pathology of the case:
Gross pathology:
The infant was 2,094 grams, 42.5 cm in crown-heel length, 28.0 cm in crown-rump length. The abdominal circumference was 26.0 cm, chest circumference was 22.5 cm, the inner intercanthal distance was 2.0 cm, and the outer intercanthal distance was 6.0 cm. The cranium was substantially enlarged but remained symmetrical. No corresponding enlargement or deformation of the face was noted. There were no other abnormal abnormal findings.
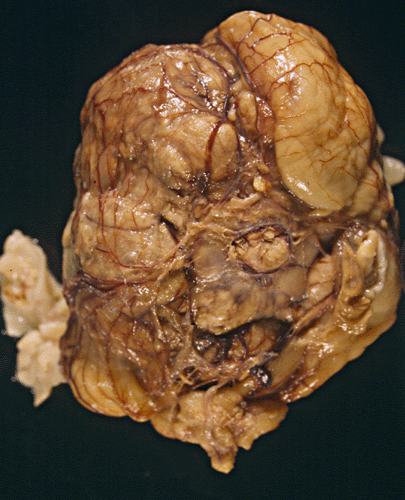
The head was enlarged to a circumference of 37.0 cm (normal range for this age is 25.0-30.5 cm). The sutures were expanded and separated. The two cerebral hemispheres appeared as two symmetrical balloon like structures. The gray and white matter were reduced to a thin mantle that varies from 0.3 to 1.0 cm in thickness (Panel A). Although some gyral markings were preserved, most of the gyral markings were compressed and displayed an "ironed out" appearance. The cerebellum was compressed and slightly flattened. The brainstem was also flattened and distorted.
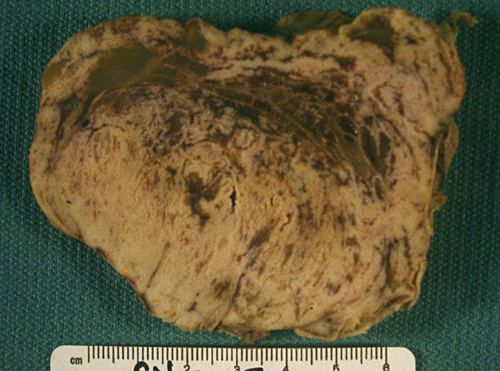
A firm to hard, centrally located, 10.0 x 8.5 x 7.0 cm tumor is located at the base of the brain (Panel A). The tumor also invaded and destroyed the central portion of the cranial base. The pituitary could not be found and the sella was entirely destroyed. The intracerebral portion of the tumor was covered by a thin membraneous tissue which distinctly demarcated the mass from the brain parenchyma. The basal ganglia and thalamus are largely destroyed by the mass. The tumor had a firm consistency on cutting. The cut surface displayed alternating vertical strands of creamy white tissue separated by thin lines of brown tissue that would suggest hemorrhage or severely congested tissue (Panel B). There was no gross evidence of necrosis or large hemorrhage. Small cystic structures containing greenish translucent gelatinous material were also present at the periphery of the tumor.
Comment: The tumor has entirely destroyed and obstructed the third ventricle and lead to the extreme hydrocephalus.
Histopathology:
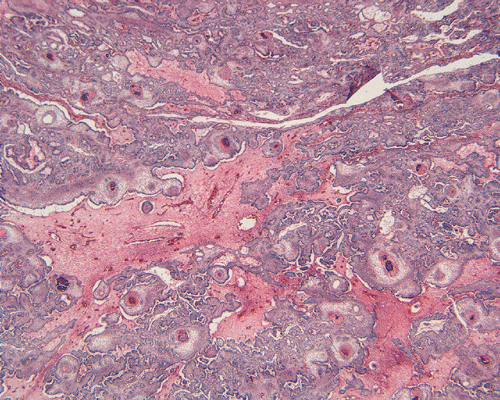
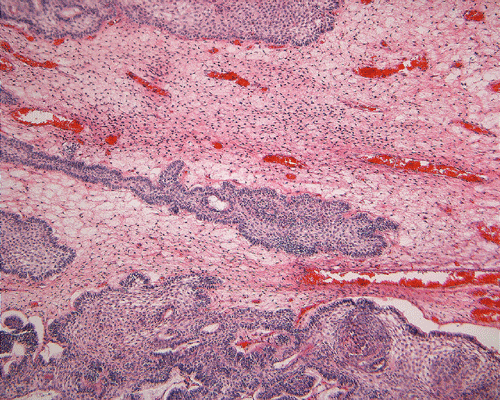
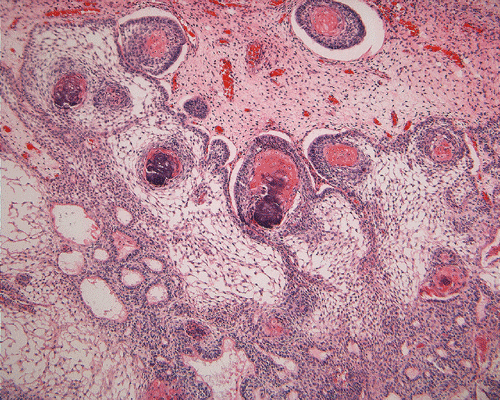
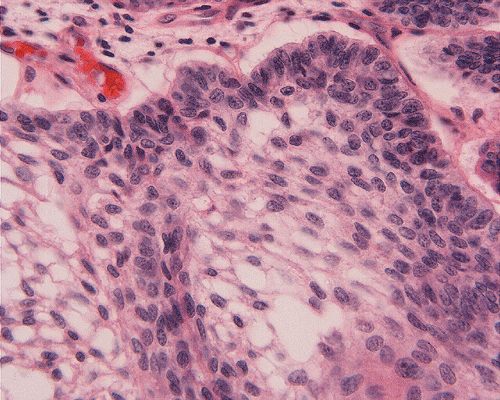
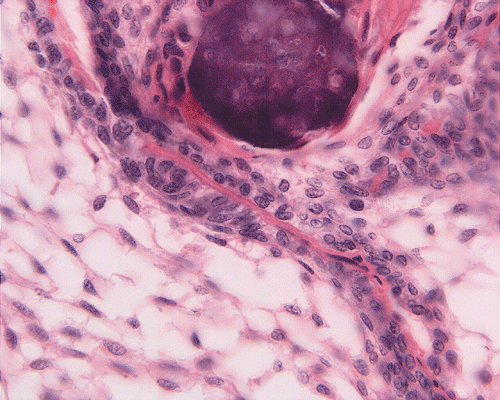
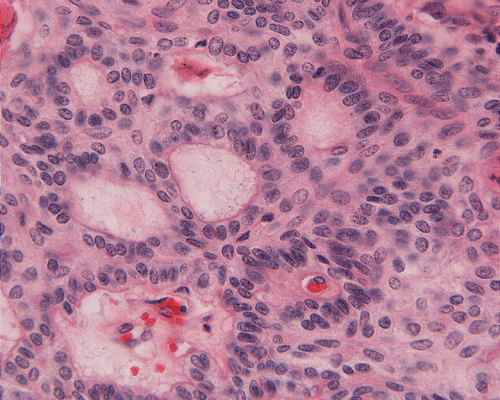
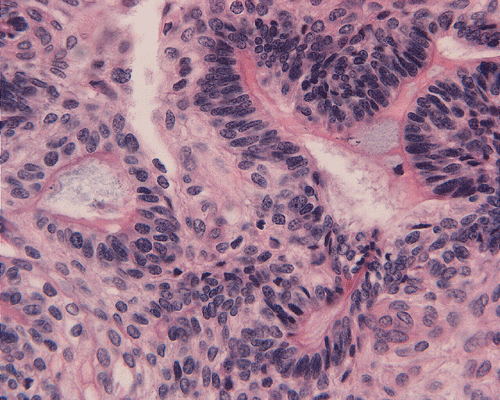
On low-magnification, the tumor appears as islands of epithelial cells that often contain a calcified core (Panel C). The tumor invades extensively into the surrounding tissue (Panel C, D, and E). The periphery of the invading islands are composed of palisading columnar cells with the center occupied by a core of stellate, mesenchyma-like cells (Panel E and F). Also within the stellate cells are calcified islands of epithelial cells. Ghost epithelial cells can be seen on higher magnification (Panel G). In some areas, the columnar cells form lumens (Panel H). In other areas, the columnar cells appear to have deposited a pink substance at the luminal border (Panel I).
| DIAGNOSIS: Congenital craniopharyngioma with extensive replacement of the brain. |
Discussion: General Information Pathology Differential diagnosis
General Information
Craniopharyngiomas are relatively uncommon tumors of the central nervous system. Biologically they are biologically benign (WHO grade I). It is, however, locally invasive and can recur 1, 2, 3. The incidence is about 0.5 to 2.5 new cases per million population per year and there is an increased incidence in Japanese children 3. They occur more frequently in children than in adults, representing 3-5% of intracranial tumors and 9 % of pediatric brain tumors. The male to female ratio is close to 1:1. About one third of cases occur in children aged 0-14 years with a peak incidence at aged 5-10 years. However, they can be seen in all ages. There is no known genetic susceptibility or association with other conditions.
Fetal and neonatal craniopharyngiomas are uncommon 1, 2, 3, 4, 5. In the series of 89 cases described by Rickert, craniopharyngiomas comprised 5.6% (5 cases) of all the fetal and neonatal tumors. Similar to many other fetal and neonatal intracranial tumors, the prognosis was grave with the longest survival of 8 weeks 4. In addition, craniopharyngiomas and teratomas usually have the largest size at the time of diagnosis and are generally detected earlier by ultrasound then other smaller lesions 4.
Craniopharyngiomas are located in the sellar-suprasellar region. About 80% of them have both a suprasellar and an intrasellar component. About 20% of the cases are exclusively suprasellar and about 5% of the cases are exclusively intrasellar 3. Clinical manifestations are due to hydrocephalus and compression and/or destruction of the sellar-suprasellar regions including the optic nerve and pituitary gland. Headache resulted from intracranial pressure secondeary to hydrocephalus occur in 60-80% of pediatric patients at presentation. Other manifestations include vomiting, nausea, visual disturbance, and diencephalic syndrome. Destruction of the pituitary gland would lead to arrested growth due to lack of growth hormone, hypothyroidism, diabetes insipidus and other hormonal deficiencies and pan hypopituitarism. Hydrocephalus is usually due to invasion of the third ventricle.
Plain skull film would show calcifications above or within the pituitary fossa and enlargement of the sella turcica. CT demonstrates mixed solid and cystic components. The solid component enhances following the administration of intravenous contrast medium 1, 2, 3.
There are two major variants: the adamantinomatous variant and the papillary variants. Grossly, craniopharygiomas usually have both solid and cystic components.
In the adamantinomatous type, the solid parts are often calcified. The cysts contain thick brownish-yellow machine oil like fluid. Cholesterol crystals can be demonstrated in unstained smear of the oily fluid under polarized light. It is important to note that the smear should not have contact with organic solvents such as xylene as the cholesterol crystals may be dissolved. The papillary variants usually appear relatively well-circumscribed and tend to lack calcifications and machine oil-like fluid 1, 2, 3, 4, 5. The adamantinomatous type tend to adhere tightly to the surrounding tissue and the papillary type is more well demarcated. The other tumor that shares the adamantinomatous histologic architecture is adamantinoma of the long bone.
Microscopically, adamantinomatous craniopharyngioma has two patterns: the ameloblastic pattern and squamous odotogenic pattern. The ameloblastic pattern is histologically indistinguishable from ameloblastoma and has morphologic features of the bell stage of the enamel organ. It is characterized by broad communicating islands and cords of stratified squamous epithelium in a system of epithelial bands and bridges. These cells are squamous in nature and include peripherally situated columanr or polygonal cells with palisading arragment at the very periphery. Enclosed within these masses are loosely arranged stellate shaped epithelial cells which closely resemble the stellate reticulum in the enamel organs. These cell islands are held together and nourished by the vascularized connective tissue stroma. The calcifying odontogenic pattern is characteristized by whorls of squamous cells with “wet” keratin and laminated keratin pearl formations with frequent central calcification. This pattern histologically is indistinguishable from calcifying odontogenic tumors of the jaws. A mixed pattern is most common in reality. Xanthogranulomatous changes with cholesterol clefts, chronic inflammation, necrotic debris and hemosiderin deposition are common features of adamantinomatous craniopharyngiomas. The xanthogranulomatous changes are highly suggestive of a diagnosis of craniopharyngioma and these changes can be extensive.
The oily fluid in adamantinomatous craniopharyngioma appears to be irritative to the surrounding glial tissue and would trigger an intense gliosis with Rosenthal fiber formation. Small biopsy tissue fragments that are obtained from these areas may suggest a pilocytic astrocytoma.
In the papillary pattern, the papillae are composed of a fibrovascular core lined by simple squamous epithelium. Keratin pearl formation is lacking. The interface of these tumors and the surrounding brain tissue is often smooth and demarcated. Papillary craniopharyngiomas must be differentiated from papillary ependymomas. The papillary pattern has no odontogenic counterpart.
Immunohistochemistry, the epithelial areas of tumor are positive for the cytokeratin and also focally positive for epithelial membrane antigen and carcinoembryonic antigen 1, 2, 3, 4. Alpha subunit of S-100 is positive in the craniopharygiomas, but beta subunit of S-100 is negative 1. Craniopharyngiomas in adults have a higher MIB-1 labeling index (Ki67) than those in children. There is no difference of MIB-1 labeling index between adamantinomatous variant and papillary craniopharyngiomas 6.
Recent studies have documented expression of beta-catenin/lymphoid enhancer factor 1 (LEF1) complex in adamantinomatous variant of craniopharyngiomas 7, 8. Mutations and over expression of b-catenin gene occur almost exclusively in the adamantinomatous craniopharyngiomas but not the papillary variants 7. Other studies have also suggest a close resemblance between craniopharyngiomas and some odontogenic tumors 9, 10. In addition, pigmented (melanin containing) adamantinomatous craniopharyngiomas have also been reported 11. Pigmented tumors have also been described in tumors of the jaws. These uncommon cases suggest possible between the cells of origin of the craniopharyngioma and jaw tumors of odontogenic origin. Both adamantinomatous and papillary types are thought to be derived from the pituitary analage from Rathke's pouch. The recent studies also suggest a lineage relationship between adamantinomatous craniopharyngioma with enamel organs and some odontogenic tumors and cysts.
Total resection and limited surgery followed by radiotherapy are two main management options for craniopharyngioma. Survival rate varied by age group, with excellent survival for patients younger than 20 years (99% at 5 years), but with poor survival for those older than 65 years (38% at 5 years) 8. The overall recurrent rate is of tumor ranging from 10% to 40% and is dependent on the nature of primary therapy, with lower rate after radical surgical removal of tumor 1, 8.
Differential diagnosis
Intracranial craniopharygiomas must be differentiated from several other lesions that would arise in the sellar-suprasellar areas.
Rathke’s cleft cyst consists of thin wall small cysts with cloudy mucinous contents lined by ciliated cubiodal or columnar epithelium. The epithelium often contains goblet cells and sometimes incorporates a metaplastic stratified squamous epithelium without keratinization or keratin pearl formation. The separation is usually straight forward. In minute specimens, it may be impossible to separate the two entities purely on morphologic grounds. A recent study shows that only 10% of craniopharyngiomas express cytokeratin 8 and are negative for cytokeratin 20. In contrast, Rathke’s cleft cyst is 100% positive for both cytokeratin 8 and 20 12. [Click here to see a case of Rathke' cleft cyst]
The intense gliosis triggered by the craniopharyngioma in the surrounding glial tissue may contain Rosenthal material. It should not be mistaken as pilocytic astrocytoma. This situation should be suspected if the radiologic diagnosis is craniopharyngioma but the tissue submitted for frozen section suggests a pilocytic astrocytoma. Additional tissue should be obtained. Separation of the two entities on permanent sections should not be a problem provided that the tissue have been thoroughly sampled and examined. [Click here to see a case of pilocytic astrocytoma]
The flattened squamous epithelium in craniopharyngiomas may suggest epidermoid cyst or dermoid cyst. This often occurs when the tissue available for examination is small. “Wet” keratin, however, is not seen in epidermoid cyst or dermoid cyst.
Papillary ependymoma may be confused with papillary craniocraniogioma. Papillary ependymoma, like other ependymomas, are positive for glial fibrillary acidic protein. Morphologically, papillary ependymoma also lacks a basement membrane and the cells processes tapers and give a coronary arrangement around blood vessels.
Xanthogranuloma of the sellar region has been suggested as a distinct clinicopathology entity by Paulus et al. Xanthogranuloma of the sellar region is described as a predominantly xanthogranulomatous tissue consisting of cholestero clefts, lympho-plasmacytic infiltration, marked hemosiderin deposition, fibrosis, foreign-body type giant cells around cholesterol clefts, eosinophilic, granular necrotic debris, and accumulation of macrophages. Focal calcifications are present in about 16% of their cases. There are small areas (less than 10% of the total area) that corresponds to calcifying odontogenic cyst in 8% of the cases. No ameloblastoma component are present. Clinically, this entity is also different from the classic adamatinomatous craniopharyngioma. The incidence in both sex are about equal. They tend to have exclusively intrasellar involvement but retrosellar involvement is less common then adamantinomatous craniopharyngioma. They are usually smaller in size but have more severe endocrinological disturbance. Xanthogranulomas also have better resectability and better prognosis 13.
Reference:
Ironside JW, Moss TH, Louis DN, Lowe JS, Weller RO. Diagnostic Pathology of Nervous System Tumours, Churchill Livingstone, 2002.
Janzer RC, Burger PC, Giangaspero F, Paulus W. Crangiopharyngioma in Pathology and Genetics- Tumours of the Nervous System. World Health Organization Classification of Tumours. By Kleihues P and Cavenee K. 2000 IARC Press page 244-246.
Guttuso P, David O, Spitz DJ, Haber, MH. Differential Diagnosis in Surgical Pathology. W.B. Saunders Company; 1st edition. 2002.
Rickert CH. Neuropathology and prognosis of foetal brain tumours. Acta Neuropathol (Berl). 1999, 98:567-576.
Bobustuc GC, Groves MD, Fuller GN, DeMonte F. Craniopharyngioma. eMedicine 2002 March. http://www.emedicine.com/neuro/topic584.htm#section~author_information
Duo D, Gasverde S, Benech F, Zenga F, Giordana MT. MIB-1 immunoreactivity in craniopharyngiomas: a clinico-pathological analysis. Clin Neuropathol. 2003, 22:229-234.
Sekine S, Sato S, Takata T, Fukuda Y, Ishida T, Kishino M, Shibata T, Kanai Y, Hirohashi S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002, 161:1997-2001.
Sekine S, Takata T, Shibata T, Mori M, Morishita Y, Noguchi M, Uchida T, Kanai Y, Hirohashi S. Expression of enamel proteins and LEF1 in adamantinomatous craniopharyngioma: evidence for its odontogenic epithelial differentiation. Histopathology. 2004, 45:573-579.
Paulus W, Stockel C, Krauss J, Sorensen N, Roggendorf W. Odontogenic classification of craniopharyngiomas: a clinicopathological study of 54 cases. Histopathology. 1997, 30:172-176.
Badger KV, Gardner DG. The relationship of adamantinomatous craniopharyngioma to ghost cell ameloblastoma of the jaws: a histopathologic and immunohistochemical study. J Oral Pathol Med. 1997, 26:349-355.
Harris BT, Horoupian DS, Tse V, Herrick MK. Melanotic craniopharyngioma: a report of two cases. Acta Neuropathol (Berl). 1999; 98:433-6.
Xin W, Rubin MA, McKeever PE. Differential expression of cytokeratins 8 and 20 distinguishes craniopharyngioma from rathke cleft cyst. Arch Pathol Lab Med. 2002;126:1174-8.
Paulus W, Honegger J, Keyvani K, Fahlbusch R. Xanthogranuloma of the sellar region: a clinicopathological entity different from adamantinomatous craniopharyngioma. Acta Neuropathol (Berl). 1999; 97:377-82.