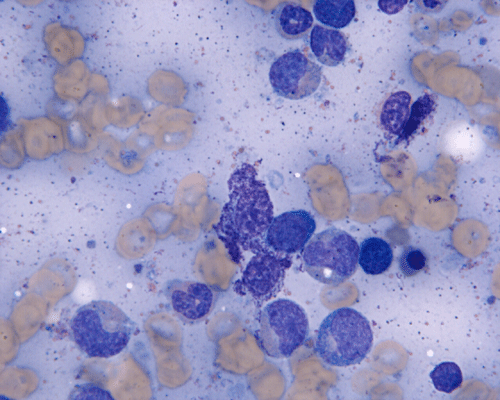
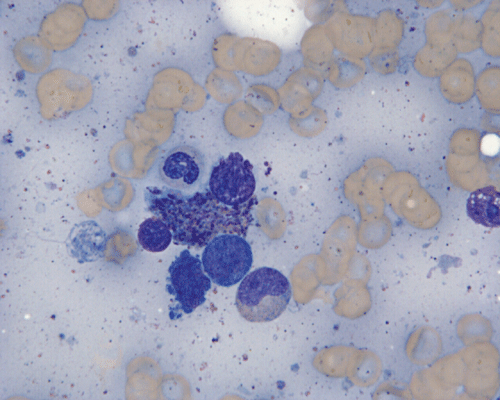
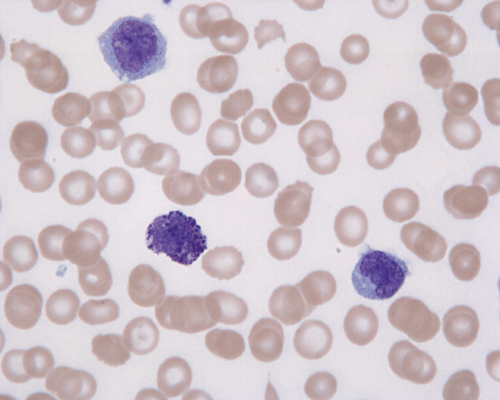
CD117
Bone marrow
DiffQuick
Bone marrow
DiffQuick
Blood
DiffQuick
| A 73 year-old Man with
Itchy Skin Rash, Hepatosplenomegaly, and Increased Level of Histamie in
Urine. September, 2005, Case 509-1. Home Page |
Robyn M. Potts, M.D., William F. Kern, M.D. Last update: April 30, 2006.
Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
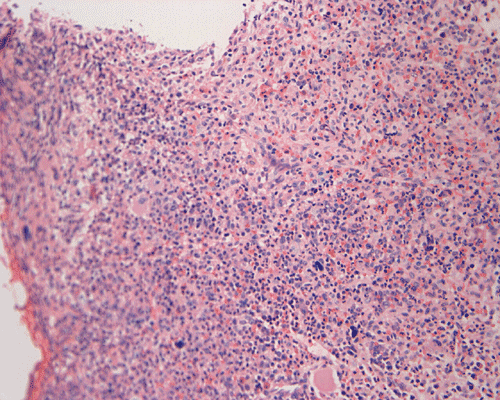
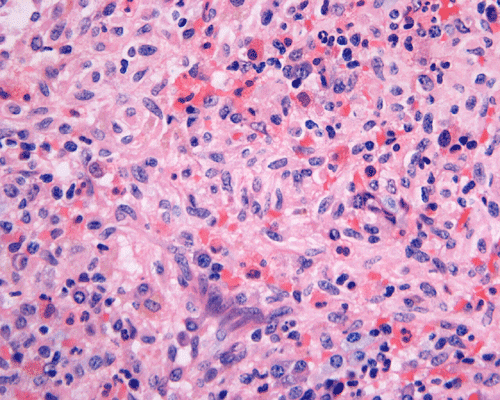
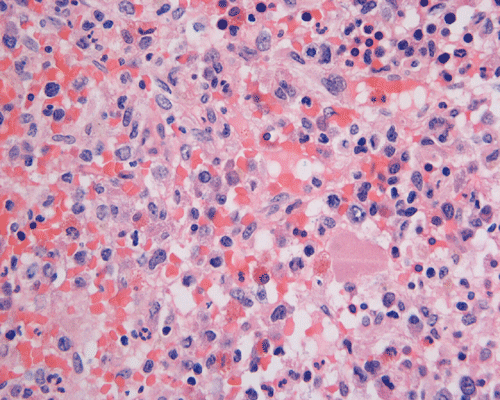
Clinical information: The patient was a 73 year-old man with an itchy skin rash, hepatosplenomegaly, and gastrointestinal bleeding. On admission, his complete blood count (CBC) showed an elevated white count (21,000/cm3) with 25% monocytes and 16% eosinophils, hemoglobin of 9.8 g/dL and 8,000 platelets/cm3. His 24-hour urine histamine was >200 mg/mL (ref 5.6-13.5 mg/mL). A bone marrow biopsy was performed and the representative photomicrographs are illustrated below:
|
|
|
|
|
|
|
|
| A. | B. | C. |
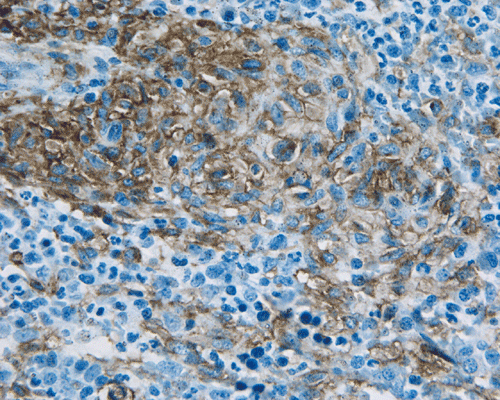
D. CD117 |
E. Bone marrow DiffQuick |
F. Bone marrow DiffQuick |
G. Blood DiffQuick |
Pathology of the case: [Click here to see a similar case]
| DIAGNOSIS: Aggressive systemic mastocytosis (WHO classification). |
Discussion: General Information Pathology Molecular Pathology
General Information:
Mast cell disease can be divided broadly into two major categories: the generally benign, skin-limited disorders typically affecting children and the systemic, clonal proliferations of mast cells in the bone marrow and other extracutaneous sites typically affecting adults 1, 2, 3.
The classification of systemic mast cell disease requires integration of clinical symptomatology, morphological assessment of the bone marrow, measurement of serum tryptase levels, and molecular/cytogenetic data. The following is the current WHO histological classification of mast cell disease 1:
In this classification, four types of systemic mastocytosis are recognized (in bold). The majority of adult patients have indolent systemic mastocytosis and a good prognosis. These patients may have symptoms of flushing, abdominal cramping, or hypotension due to systemic release of mast cell granule contents. If the patient has an associated non-mast cell clonal hematopoetic disorder, the prognosis is typically dictated by the non-mast cell clonal hematopoetic disorder, which is most commonly chronic myelomonocytic leukemia. Cytopenias and life-threatening organomegaly characterize aggressive systemic mastocytosis. Rarely patients present with or evolve to a frank mast cell leukemia.
The patient in the presented case was diagnosed with aggressive systemic mastocytosis, with documented bone marrow, liver and spleen involvement, and succumbed to his disease about two months after the presented biopsy.
Pathology and Diagnostic Criteria
The diagnosis of mast cell disease according to the World Health Organization criteria can be rendered if either one of the major and one of the minor criteria or three minor criteria are met which are listed below 1.
|
Table 1. WHO Criteria for Diagnosis of Systemic Mastocytosis |
|
Major Multifocal, dense infiltrates of mast cells (15 or more mast cells in aggregates) detected in sections of bone marrow and/or other cutaneous organ(s), and conforimed by tryptase immunohistochemistry or other special stains.
|
|
Minor
The diagnosis of systemic mastocytosis may be made if one major and one minor criterion are present, or, if three minor criteria are fulfilled. |
Since increased mast cells can be seen in a variety of benign and reactive conditions in the bone marrow. The key morphological features indicating non-reactive, clonal proliferation of mast cells are the abnormal aggregation of mast cells in bone marrow or extracutaneous sites and/or the recognition of atypical mast cells. Mast cell disease should be considered in the differential of cases characterized by extensive myelofibrosis and may be found in a perivascular or trabecular arrangement. As the histological findings can be subtle, knowledge of appropriate clinical information and a high index of suspicion is necessary to identify mastocytosis.
Normal mast cells have a round or oval, centrally located nucleus with uniform distribution of dark purple granules in the cytoplasm. Mast cells are well demonstrated by toludine blue and Giemsa stains, and also by immunohistochemistry for CD 117 or tryptase. Napthol ASD chloracetate esterase also highlight mast cells specifically. Mast cells are also positive for CD45, CD33, and CD68 but these markers are less specific than tryptase. Bone marrow aspirations stained with Giemsa are most useful for recognizing atypia in mast cells. It should, however, note that Giemsa stain may be false negative in formalin fided tissue. Hallmarks of atypia in mast cells include spindling of mast cells, hypogranulation, uneven granule distribution, and nuclear lobulation. Recognition of the so-called mast cell granuloma, in which mast cell aggregrates are associated with lymphocytes and eosinophils can be helpful.
In the bone marrow, three morphologic patterns of mast cell accumulation have been recognized. Type I shows focal clusters of mast cells with preservation of bone marrow fat and other hematopoetic elements. The burden of mast cells is more in Type II and usually shows patchy clusters of mast cell replacing bone marrow in a hypercellular marrow background with osteosclerosis and fibrosis. Type III is diffuse replacement of the marrow space with mast cells, often with circulating mast cells in peripheral blood. It is also important to note that mast cells are easily over looked on routine H and E stain or are mistaken for histiocytes.
By flow cytometry, aberrant expression of CD 25 and CD 2 has been used to define an atypical population of mast cells. About 95% of systemic mast cell disease cases aberrantly express CD 25 and 70% aberrantly express CD 2.
Molecular Alterations in Systemic Mast Cell Disease
A well defined point mutation is present in the KIT gene in most patients with systemic mastocytosis. The most common mutation in systemic mast cell disease is an activating (gain of function) mutation in the KIT gene codon 816 (D816V). Interestingly, this mutation is in the tyrosin e kinase domain and interfere binding of imatinib mesylate (Gleevac) to the enzymaztic site of the Kit receptor protein, rendering these patients resistant to this drug. The KIT 816 mutation occurs in approximately 80% of systemic mast cell disease patients. Another 5% of the patients with systemic mast cell disease have other mutations in the KIT gene. A novel form of mutation (Phe522Cys, in the transmembrane portion of the Kit receptor protein) which allows treament with Imatinib mesylate (Gleevac) has been described 4.
A second important cytogenetic alteration found in a subset of systemic mast cell disease patients is an interstitial deletion on Chromosome 4 which causes a fusion of the FIP1-like 1(FIP1L1) and PDGRFA proteins 5. This deletion is detectable by FISH and has been associated with patients with eosinophilia and mast cell disease as well as patients with the hypereosinophilic syndrome with out mast cell disease.
Finally, in patients with associated clonal non-mast cell disorders, the bone marrow cytogenetics are usually characteristic of the non-mast cell disorder.
Reference: