
| A 54 year-old Man with
Swelling Between his Eyes. September, 2008, Case 809-1. Home Page |
Vladislav Zakharov, M.D. 1, Cheng Z. Liu, M.D., Ph.D., M.D. 1, Jignesh M. Modi, M.D. 2, Kar-Ming Fung, M.D., Ph.D. 1 Last update: September 2, 2008.
1 Department of Pathology and 2 Department of Radiology University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Clinical information: The patient was a 54 year-old man who came to our clinic with the chief complain of a swelling between his eyes for 18 months. He reported frequent headache, changes in vision including double vision and decreased ability to smell. There was also minor changes in taste and dizziness but he did not report any syncope events. Physical examination revealed widening of nasal bridge and hypertelorism. His facial expression, motility, and strength are preserved. The pupils are equal, round and reactive to light. A CT scan and MRI was performed and demonstrated a large cystic mass as illustrated below. A biopsy was performed through the nasal cavity which yielded small fragments of spindle cell neoplasm with no high grade features demonstrated. Based on the biopsy result, the mass was excised and yielded the following representative images.
|
|
|
|
|
|
|
| A | B | C | D | E | F |
|
|
|
|
|
|
|
| G | H | I | J | K | L |
|
|
|||||
| Scanned slide |
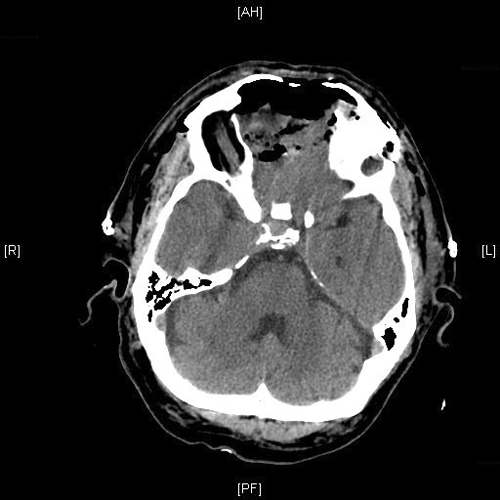
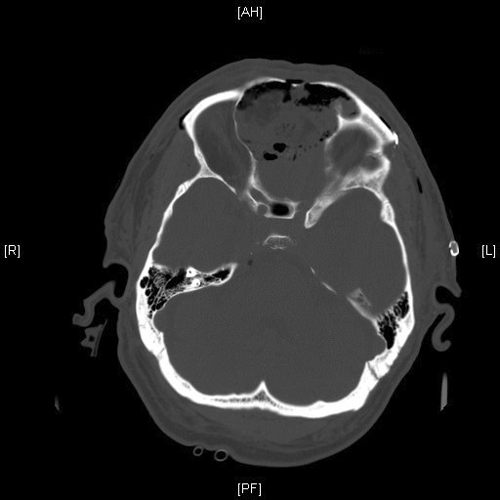
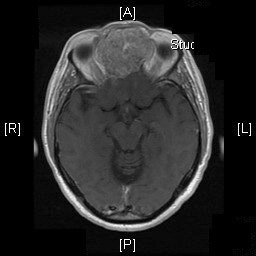
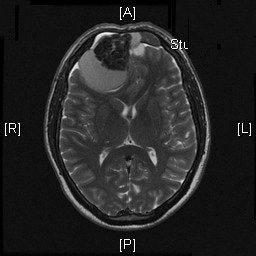
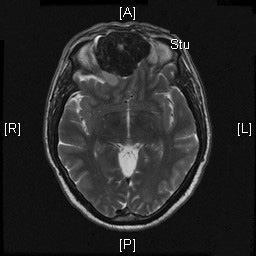
Radiologic Images of the Case: CT scan demonstrated a large midline mixed density mass with fat density that occupies the ethmoid sinus with extension through the cribiform plate into the inferior portion of the anterior cranial fossa. The mass is 5.1 x 3.2 cm in greatest dimension (Panel A). Bone windows of CT scan demonstrated bone expansion suggestive of remodeling (Panel B). On MRI, spoiled gradient recalled (SPGR ) post contrast images showing mass is either avidly enhancing or bright on T1 precontrast. (Panel C). The mass hypointense signal intensity compared to brain parenchyma with surrounding areas of increased signal (suggestive of entrapped mucus) on T2 weighted images (Panel D and E).
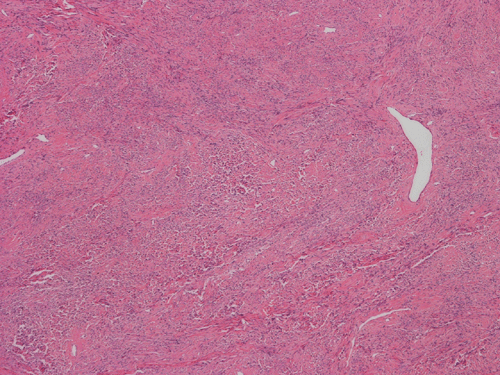
Pathology of the Case: The excised specimen is a 61 gram, 5.4 x 4.3 x 4.2 cm solid, rubbery to firm, basically round nodules. The external surface is smooth and glistening in about 70% of the areas with the remaining areas adhered to small amount of fibrous tissue. The cut surface is pink to tan with a whorled pattern. There was no hemorrhage or necrosis. In some areas, the cut surface is a little gritty and contains small opaque dots, 0.1-0.2 cm in greatest dimension, that are suggestive of calcification.
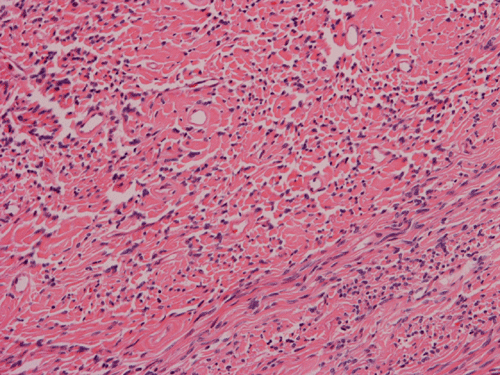
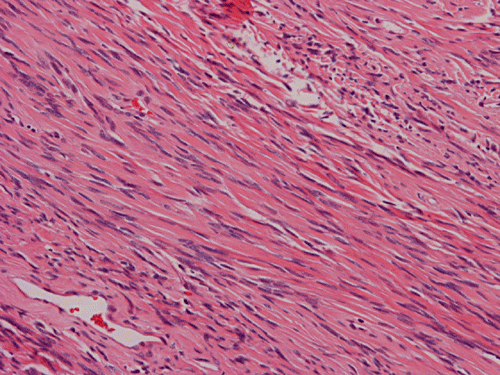
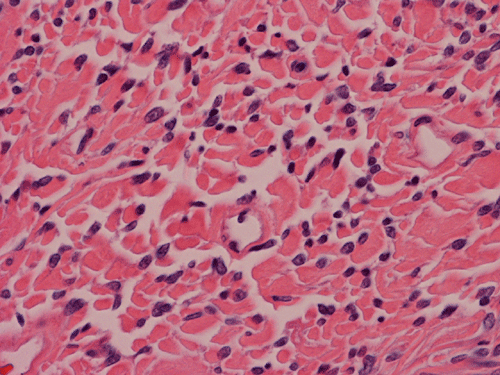
Histologically, the tumor is composed predominantly of strands of collagen fibers arranged in interlacing bundles (Panel F, G, H, I, J ). The histologic pattern is rather homogeneous among different areas and no area with particular hypocellularity or hypercellularity are found (Panel F). In between the collagen fibers are cigar shaped nuclei without high grade features (Panel J). The nuclei and the collagen fibers maintain an interesting regularly randomized relationship reminiscent of a checker board pattern (Panel I). A few small fragments of entrapped, mature bone are also found within the tumor. Mitoses and necrosis are not seen.
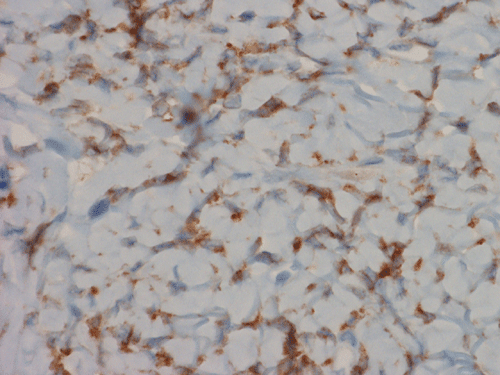
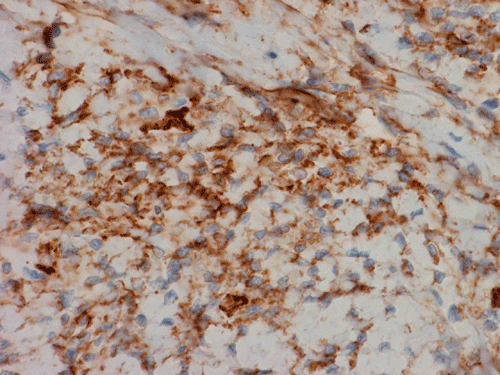
Immunohistochemistry demonstrated positive immunoreactivity in tumor cells for Vimentin, Bcl2 (Panel K) and CD34 (Panel L) but not CD99. The tumor cells are negative for muscle specific actin, desmin, S100, cytokeratin AE1/AE3, and epithelial membrane antigen (EMA).
| DIAGNOSIS: Solitary fibrous tumor |
Discussion:
General Information
The identification of solitary fibrous tumor (SFT) as a distinct entity is generally credited to Klemperer and Rabin 1, who first described five cases back in 1931. SFT is an uncommon mesenchymal tumor 2 originally considered almost exclusively located in the pleural cavity. In the earlier years, it was also known as localized fibrous mesothelioma. SFT has now been reported to have occurred at many extra thoracic locations, including mediastinum, lung, prostate, kidney, thyroid, and the spinal cord 3.
SFTs typically occur in middle-aged adults between 20 and 70 years with no sex predilection. Occasional cases occur in children and adolescents. Although SFTs are generally indolent neoplasms that are cured with complete surgical resection, histologically and clinically malignant cases have been reported in 10% to 15% of thoracic SFTs 4. Malignant transformation may also occur within histologically benign SFTs even after several years of diagnosis 5.
Most SFTs of soft tissue present as well delineated, slowly growing, painless masses. Depending on the location, especially in the nasal cavity, the orbit and the meninges, large tumors may give rise to manifestations related to compression or obstruction. About 5% of the cases of SFT, particularly those located in the pelvis and retroperitoneum, are associated with hypoglycemia which related to the production of an insulin-like growth factor 7.
Excision is currently considered a standard of care. Adjuvant chemotherapy or radiotherapy may be considered in malignant variants. Although most cases are benign, the behavior of SFT can be unpredictable based on histologic features alone. There is no strict correlation between morphology and clinical behavior. Roughly, 10 to 15% behave aggressively 4, thus long-term follow-up is mandatory. Lesions located in the mediastinum, abdomen, pelvis, and/or retroperitoneum also tend to behave more aggressively than those in the limbs. Metastases are most frequently observed in lungs, bone and liver.
Pathology
Grossly, SFTs are well circumscribed, often partially encapsulated masses, measuring between 1 and 25 cm. The pleura is still the most common site. Tumors arising from the parietal pleura are more difficult to be resected than those from the visceral pleura because of their size and adhesion to the chest wall requiring extrapleural resection 5. On section, tumors compose of multiple confluent nodules with whitish and firm appearance; myxoid and hemorrhagic changes are occasionally observed. Tumor necrosis and infiltrative margins are signs of malignancy and considered to be suggestive of aggressive tumor behavior.
Microscopically, SFTs show a wide range of morphological features but hyalinized collagenous fibrous tissue is a prominent component. Many SFTs have branching blood vessels like those found in hemangiopericytomas. The distinction between hemangiopericytoma and solitary fibrous are becoming blurred and these two entities are often discussed as one entity (hemangiopericytoma-solitary fibrous tumor) in several major text 3, 6, 8 . Hemangiopericytoma, giant-cell-rich (previously called giant cell angiofibroma) and the fat-forming (previously called lipomatous hemangiopericytoma) are considered to be variants of hemangiopericytom-solitary fibrous tumor.
As reflected by their name, SFTs are fibrous and contain large collagenized areas and vessels with thick and/or hyalinized walls. In the classic cases, the collagenous fibrous component is strikingly hyalinized.The cells usually have low grade, elongated, cigar shaped nuclei and they often insinuate among collagen fibers. For those arising in the pleura, they are often covered by mesothelial cells. Entrapped alveolar or bronchial epithelial component at their serosal margin may be suggestive of an epithelial component of these tumors.
Some tumors may be more cellular and less fibrous. SFTs have often been referred as having a ‘patternless’ growth pattern. In fact, two partially overlapping patterns are recognizable in most of the SFTs. The more common form, so called fibrous SFT, is characterized by alternating hypercellular and hypocellular fibrous areas. The cells in hypercellular areas are predominantly of round-to-spindle cells with a fascicular, storiform or fibrosarcoma-like arrangement. One of the most consistent features of the fibrous form of SFT is the presence of numerous, medium-sized, branching vessels with characteristically thickened and hyalinized walls. The second form, a cellular form, of SFT resembles what had been called hemangiopericytoma prior to 1990 7. As opposed to the more common fibrous form, cellular SFTs usually have a monotonous appearance, even, moderate to high cellularity, little intervening fibrosis, numerous thin-walled ‘staghorn’ branching vessels, and round-to-oval monomorphic tumor cell nuclei. They are usually large and tend to be found in the deep soft tissues of the thigh, pelvis retroperitoneum, limb girdles, head and neck, as well as in the meninges. Myxoid change, areas of fibrosis and interstitial mast cells are commonly observed in SFTs. Mitoses are generally scarce, rarely exceeding 3 mitoses per 10 high-power fields. Some SFTs may contain mature adipocytes and/or giant multinucleated stromal cells, overlapping morphologically with the so-called lipomatous haemangiopericytoma and giant cell angiofibroma.
CD34 is expressed in about 90% of the pleural cases. Other molecular markers including Bcl-2, CD99, epithelial membrane antigen (EMA), and actin have also been demonstrated to be positive, but cytokeratin, SMA, desmin, S-100, EMA, HMB-45, and c-kit are usually negative 9.
The criteria for malignant or aggressive behavior vary among different studies. The malignant variants of SFTs are usually hypercellular lesions with at least focally moderate to marked cytological atypia, tumor necrosis, increase in mitotic activity (4 or more mitoses per ten high-power fields) and/or infiltrative margins. According to Gold et al., 10 large tumor size (> 10 cm) and the presence of a histologically malignant component, as defined by an areas of increased cellularity with a mitotic index >4 mitoses per 10 high-power fields and the lack of alternating sclerotic hypocellular areas (observed in 20% of SFT), are the most important predictors of poor outcome. Rare cases that show abrupt transition from conventional benign-appearing SFT to high grade sarcoma most likely represent malignant transformation. Secondary malignant transformation of SFTs has been associated with diminished CD34 expression, positive p53 immunoreactivity, and high expression of Ki-67. 9, 11 Several genetic aberrations have been identified but no consistent genetic markers have been defined. 12, 13, 14, 15, 16
Differential diagnosis
SFT is a spindle-cell neoplasm and has a wide differential diagnosis. In soft tissues, this lesion should be distinguished from other spindle-cell neoplasm such as schwannoma, myofibroblastoma, metastasis from spindle-cell carcinoma, low-grade fibromyxoid sarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor and others.
Reference
Klemperer P, Rabin C B. Primary neoplasm of the pleura: a report of five cases. Arch Pathol. 1931 11:385–412.
el-Naggar A, Ro J, Ayala A, Ward R, Ordóñez N. Localized fibrous tumor of the serosal cavities. Immunohistochemical, electron-microscopic, and flow-cytometric DNA study. Am J Clin Pathol 1989 92:561–5.
Fletcher CDM. The evolving classification of soft tissue tumors: an update based on the new WHO classification. Histopathology 2006 48:3–12.
Vallat-Decouvelaere A, Dry S, Fletcher C. Atypical and malignant solitary fibrous tumors in extrathoracic locations: evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol 1998 22:1501–11.
de Perrot M, Kurt A-M, Robert JH, Borisch B, Spiliopoulos A. Clinical behavior of solitary fibrous tumors of the pleura. Ann Thorac Surg 1999 67:1456.
Weiss SW, Goldblum JR. Soft tissue tumors of intermediate malignancy of uncertain type. In Enzinger and Weiss’s Soft Tissue Tumors. Page 1093-1160. 5th Edition, 2008 Mosby.
Nagase T, Adachi I, Yamada T, Murakami N, Morita K, Yoshino Y, Katayanagi K, Kurumaya H. Solitary fibrous tumor in the pelvic cavity with hypoglycemia: report of a case. Surg. Today 2005 35:181–184
Gengler C, Guillou L. Solitary fibrous tumor and haemangiopericytoma: evolution of a concept. Histopathology 2006 48:63–74.
Yokoi T, Tsuzuki T, Yatabe Y, Suzuki M, Kurumaya H, Koshikawa T, Kuhara H, Kuroda M, Nakamura N, Nakatani Y, Kakudo K. Solitary fibrous tumor: significance of p53 and CD34 immunoreactivity in its malignant transformation. Histopathology 1998 32:423–32.
Gold JS, Antonescu CR, Hajdu C, Ferrone CR, Hussain M, Lewis JJ, Brennan MF, Coit DG. Clinicopathologic correlates of solitary fibrous tumor. Cancer 2002; 94; 1057–1068.
Brozzetti S, D'Andrea N, Limiti M, Pisanelli M, De Angelis R, Cavallaro A. Clinical behavior of solitary fibrous tumors of the pleura. An immunohistochemical study. Anticancer Res 2000 20:4701–6.
Horton ES, Dobin SM, Donner LR. A clonal t(8;12)(p11.2;q24.3) as the sole abnormality in a solitary fibrous tumor of the pleura. Cancer Genet Cytogenet 2007 172:77-79.
Dal Cin P, Pauwels P, Van Den Berghe H. Solitary fibrous tumour of the pleura with t(4;15)(q13;q26). Histopathology. 1999 35:94-5.
Havlik DM, Farnath DA, Bocklage T. Solitary fibrous tumor of the orbit with a t(9;22)(q31;p13). Arch Pathol Lab Med. 2000 124:756-8.
Debiec-Rychter M, de Wever I, Hagemeijer A, Sciot R. Is 4q13 a recurring breakpoint in solitary fibrous tumors? Cancer Genet Cytogenet. 2001 131:69-73.
Dal Cin P, Sciot R, Fletcher CD, Hilliker C, De Wever I, Van Damme B, Van den Berghe H. Trisomy 21 in solitary fibrous tumor. Cancer Genet Cytogenet. 1996 86:58-60.
Cases of the Month Evaluation Coordinator: KarMing-Fung@ouhsc.edu
Copyrights reserved.