

Squash preparation
Squash preparation
Squash preparation

Frozen Section
Frozen Section
Alcian blue-PAS
EMA
AE1/AE3
Semithin
| A 51 year-old Man with a Sellar Mass. April, 2012, Case 1204-1. Home Page |
Kwok Ling Kam, M.B., B.S., FRCPA 1, Kar-Ming Fung, M.D., Ph.D.2 Last updated on on May 25, 2020.
1 Department of Pathology, Feinberg School of Medicine, Northwestern University, Chicago, Illinois
2 Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma
Clinical information:
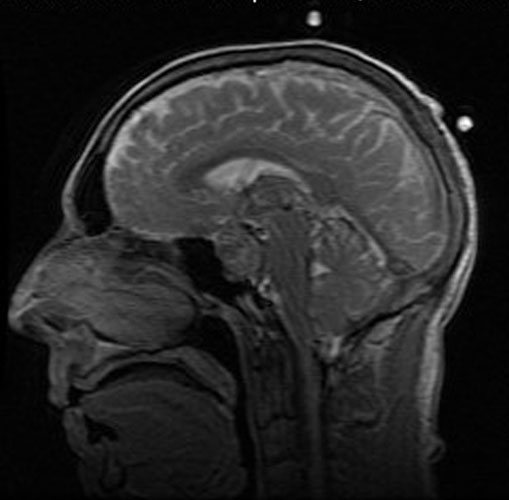
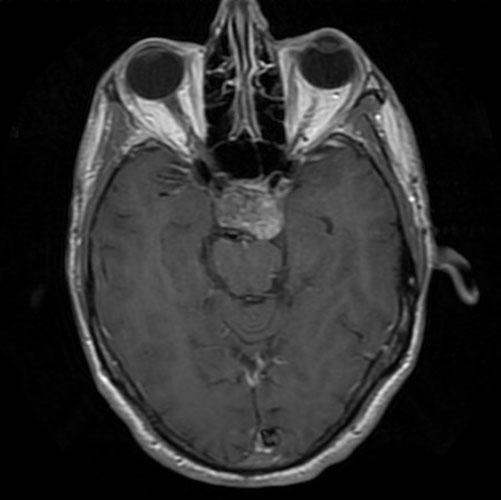
The patient was a 51 year-old man who was referred to this institute for treatment of a sellar mass. MRI revealed a 2.7 x 1.9 cm mass in the sellar region suggestive of a macroadenoma. A transphenoidal resection was performed and yielded the following images.
|
|
|
|
|
|
|
| A |
B Squash preparation |
C Squash preparation |
D Squash preparation |
||
|
|
|
|
|
|
|
|
E Frozen Section |
F Frozen Section |
G | H | I |
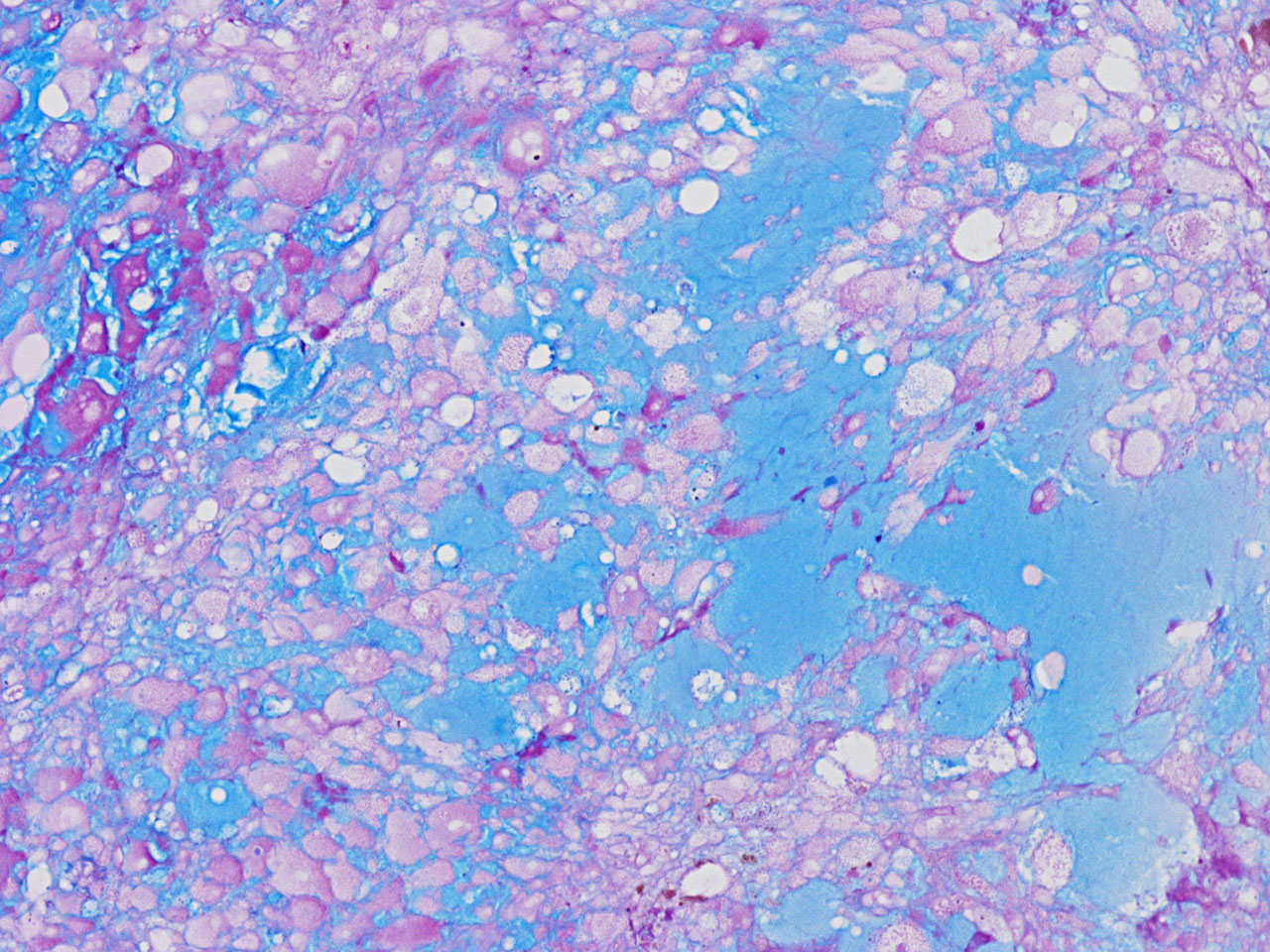
J Alcian blue-PAS |
|
|
|
|
|||
|
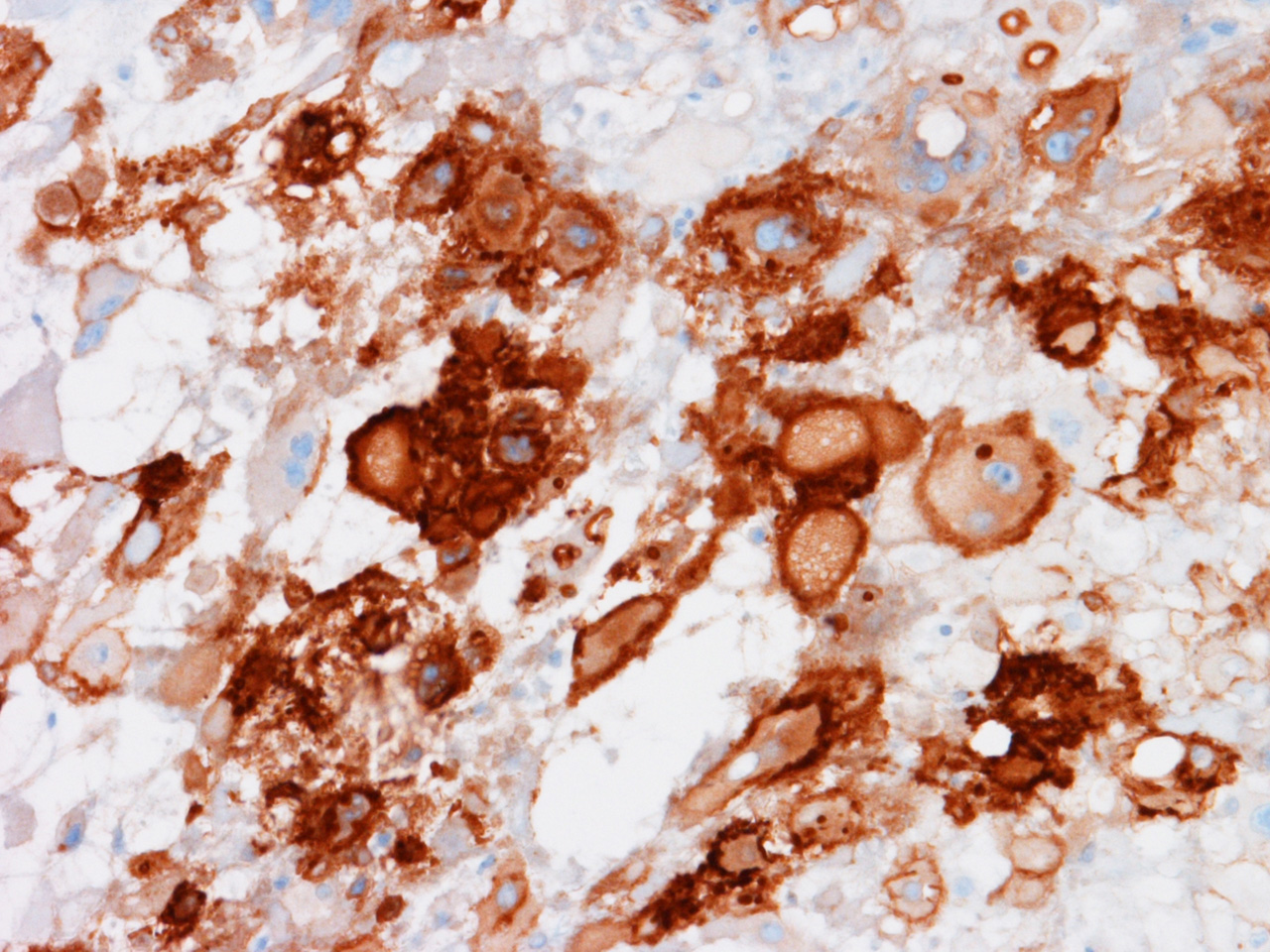
K EMA |
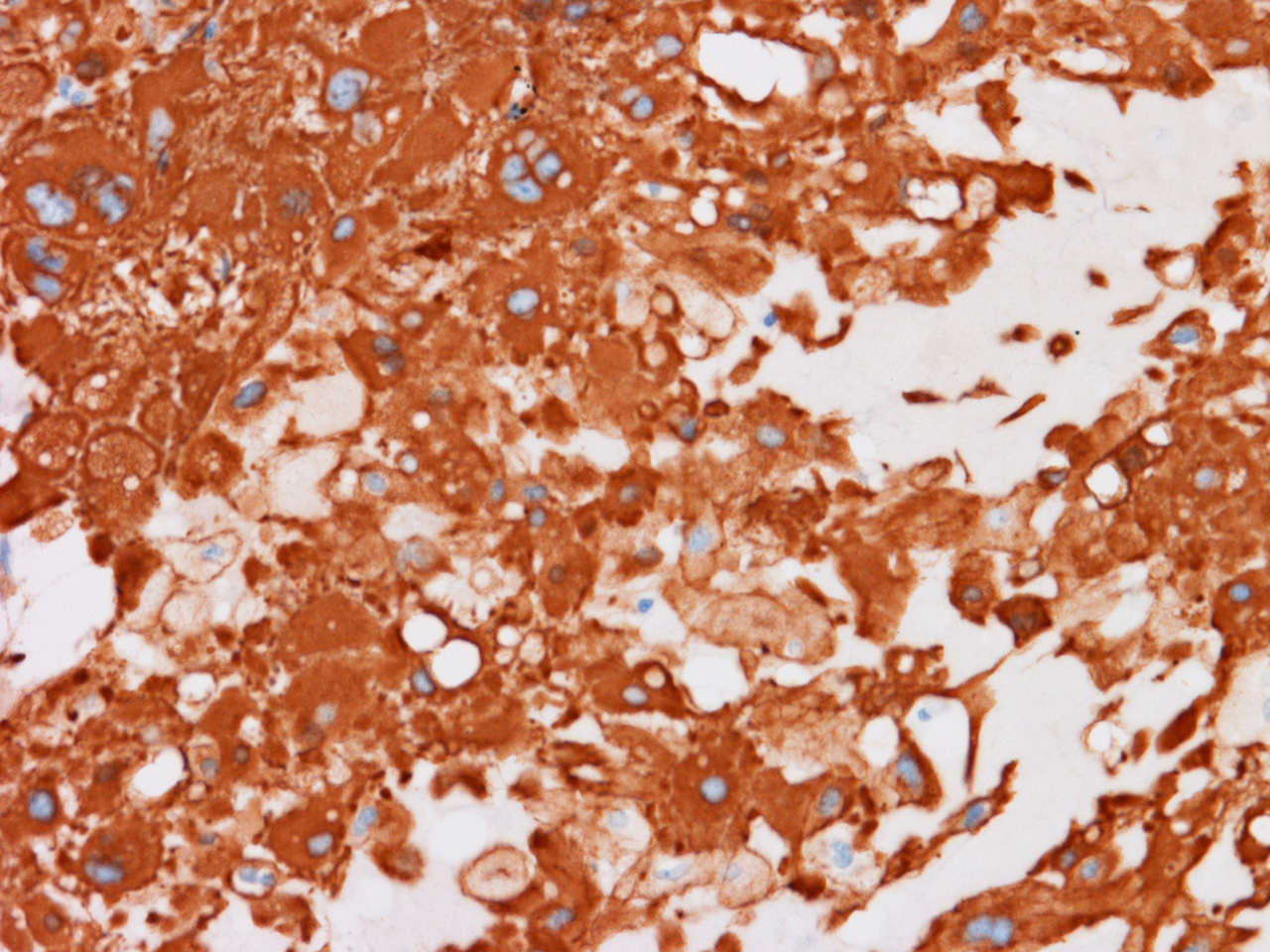
L AE1/AE3 |
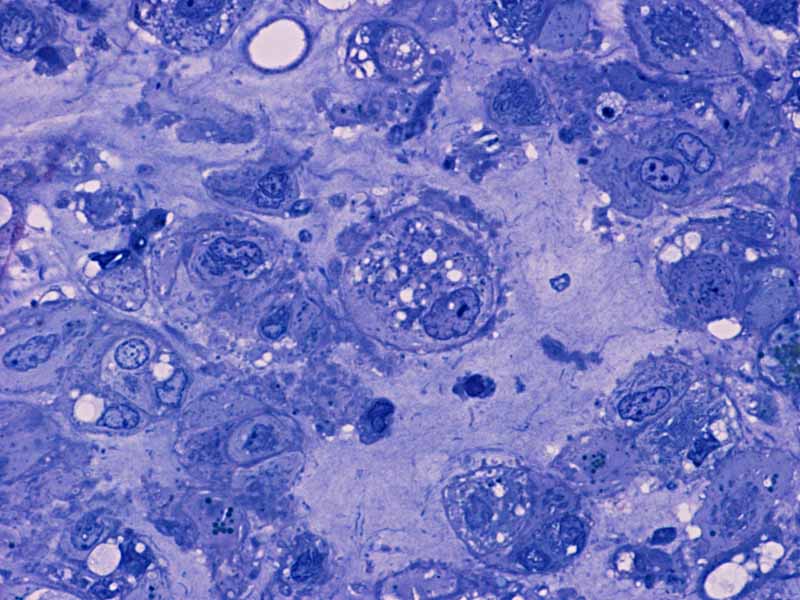
M Semithin |
Pathology of
the Case: The MRI images
clearly indicate the location of the lesion is in the sellar region with
expansion of the sellar. Radiographically, the lesion is well-circumscribed and
most consistent with a macroadenoma. The histologic details of the permanent
section can be viewed in that online slide (Panel A).
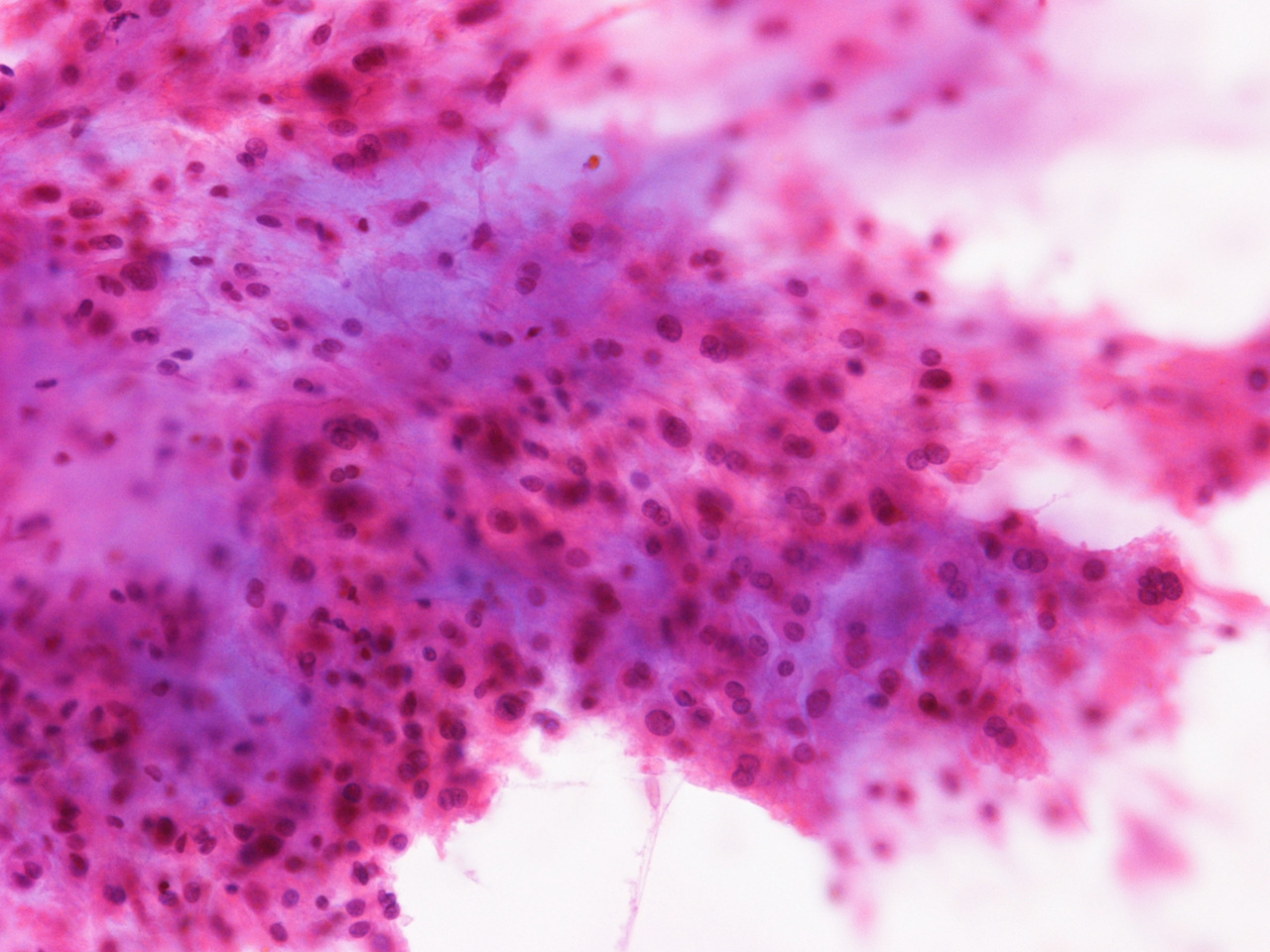
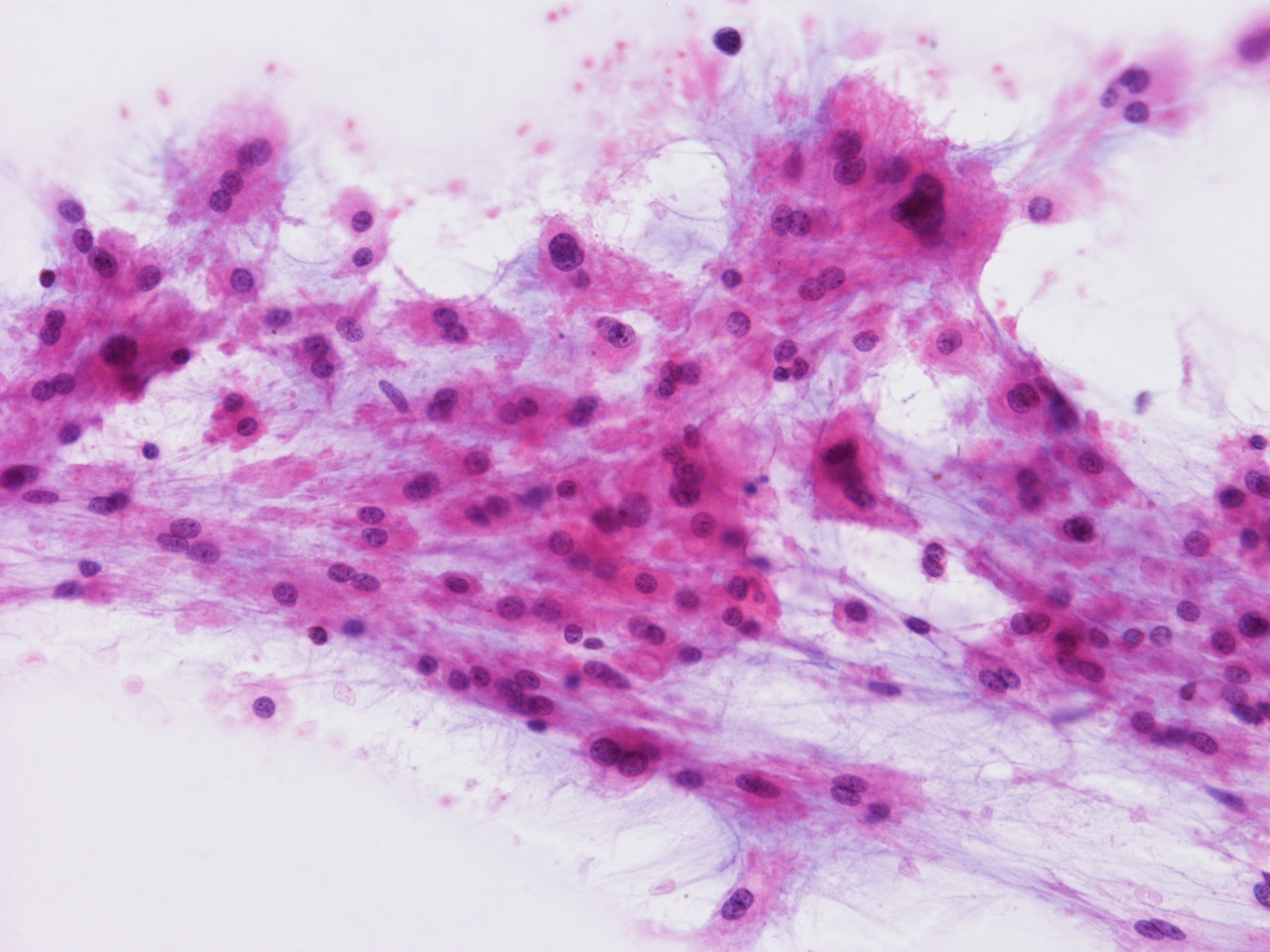
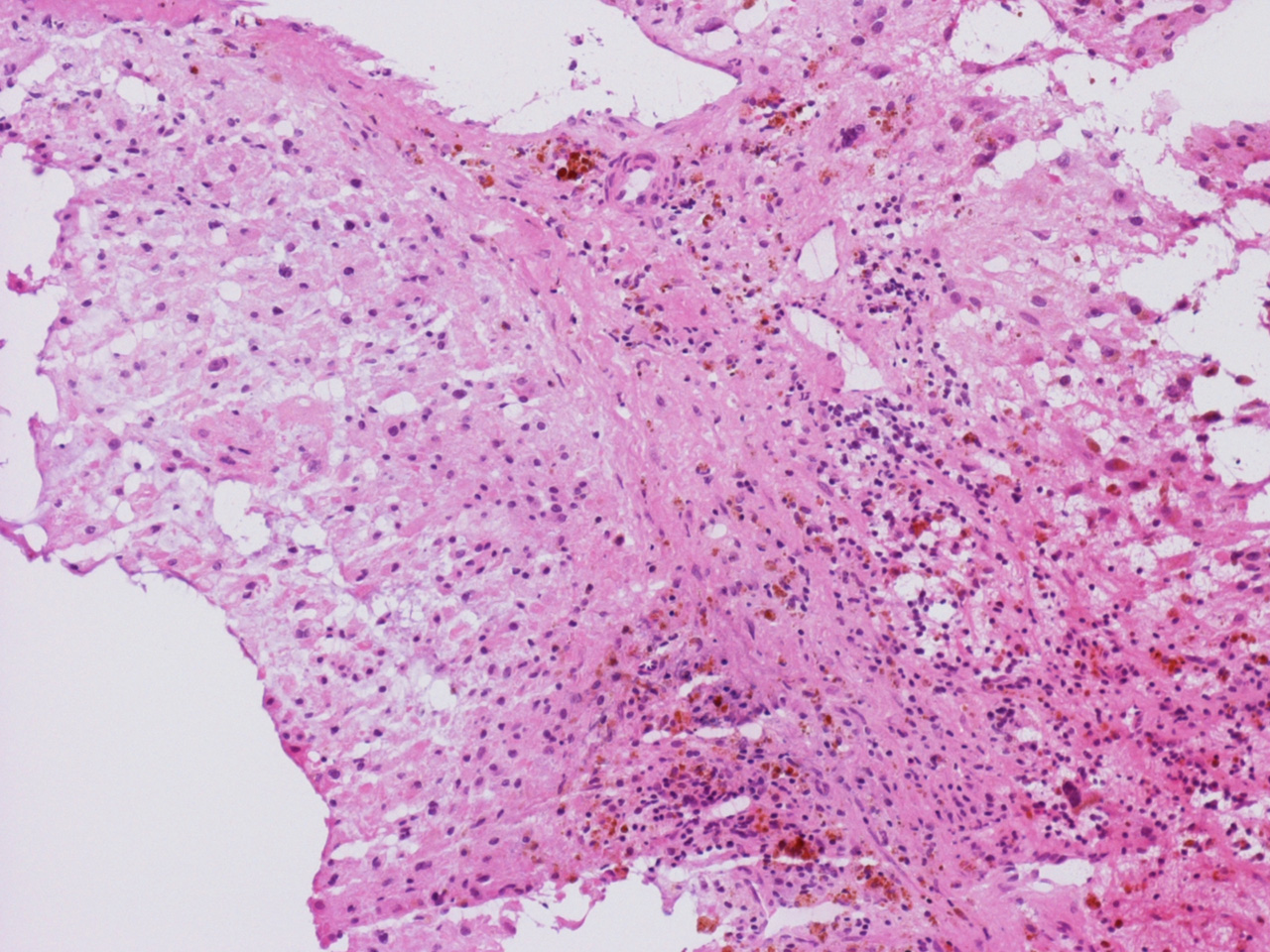
At the time of operation, a cytologic squash preparation (H&E) was prepared (Panels B, C, and D). On low magnification, the lesion is composed of large clusters of
eosinophilic cells with centrally located nuclei. There is some bluish acellular
substance admixed with the tumor cells (Panel B). If you pay attention, some of the cells are arranged in short
chains (arrows in Panel C). This is a frequently seen phenomenon in chordoma. On high
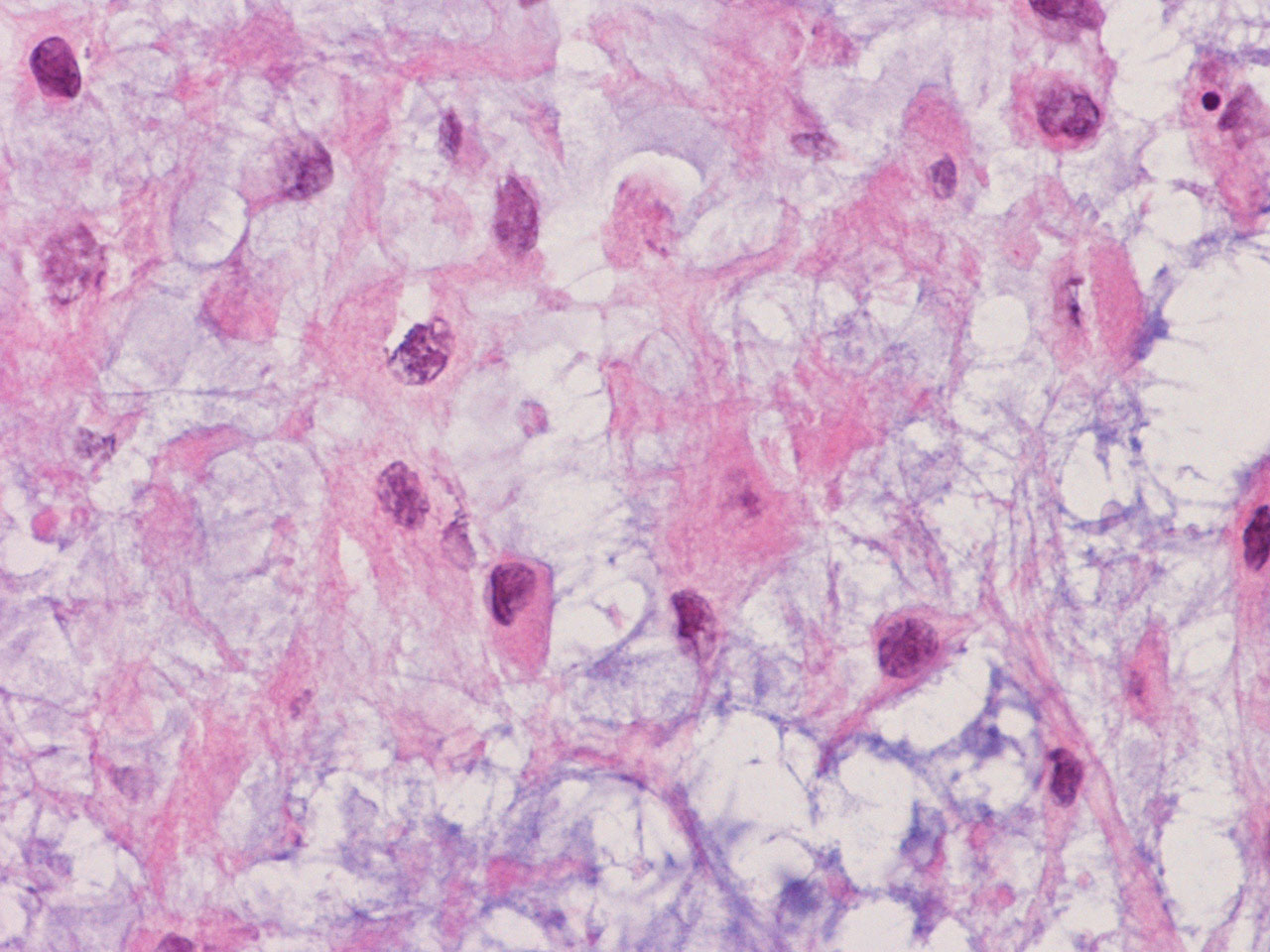
magnification, the cells have centrally located medium sized to large,
hyperchromatic nuclei. The cytoplasm is finely eosinophilic but not particularly
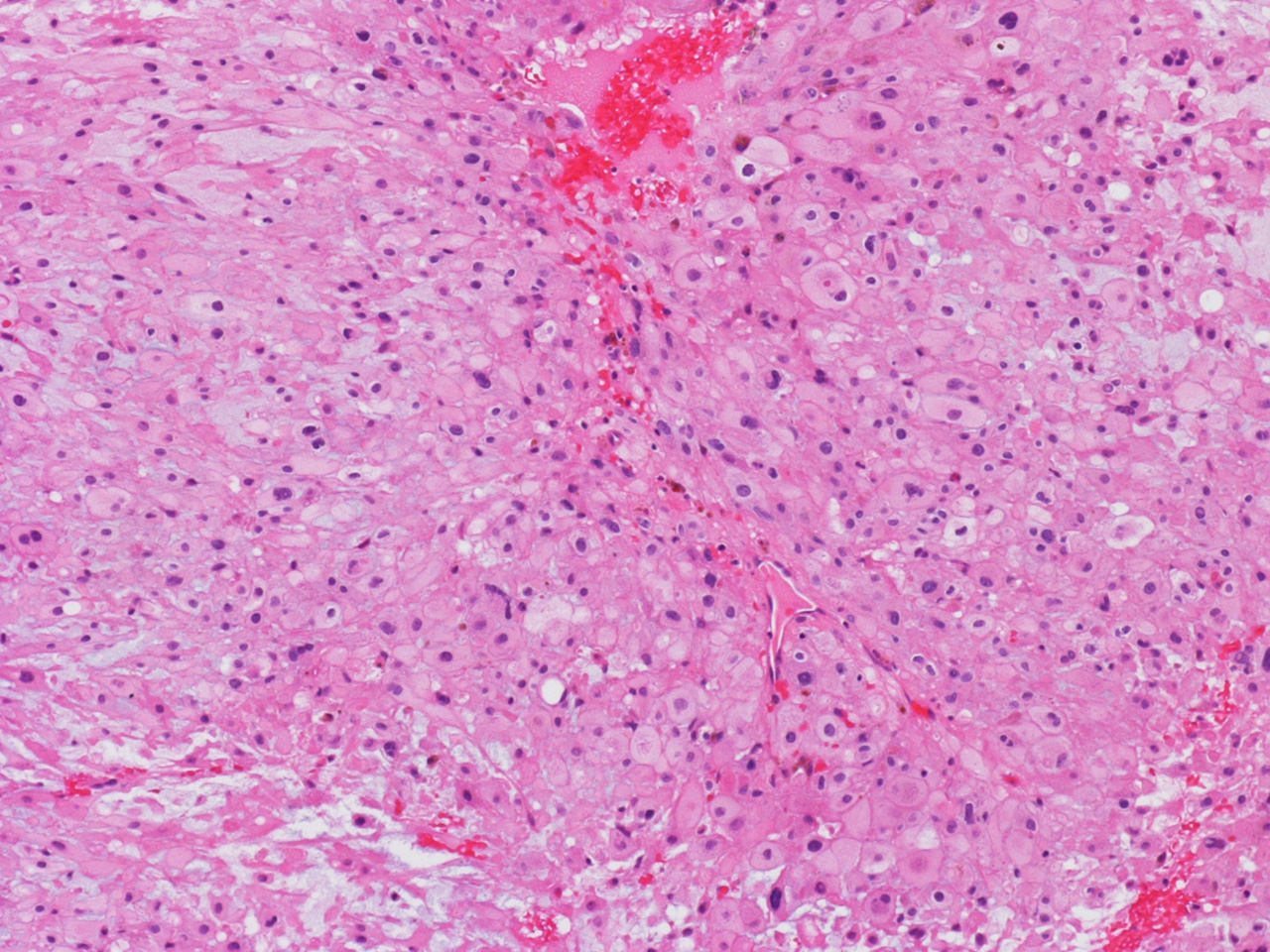
bubbly (Panel D). The frozen sections (Panels E and F) reflect the cytologic features. The tumor is composed of solid
sheets of large tumor cells admixed with small amount of fibrous areas, mild
chronic inflammatory cell infiltration and hemosiderin depositions (Panel E). On high magnification, the tumor cells are admixed with bluish
extracellular material. The cytoplasm is coarsely granular with fine
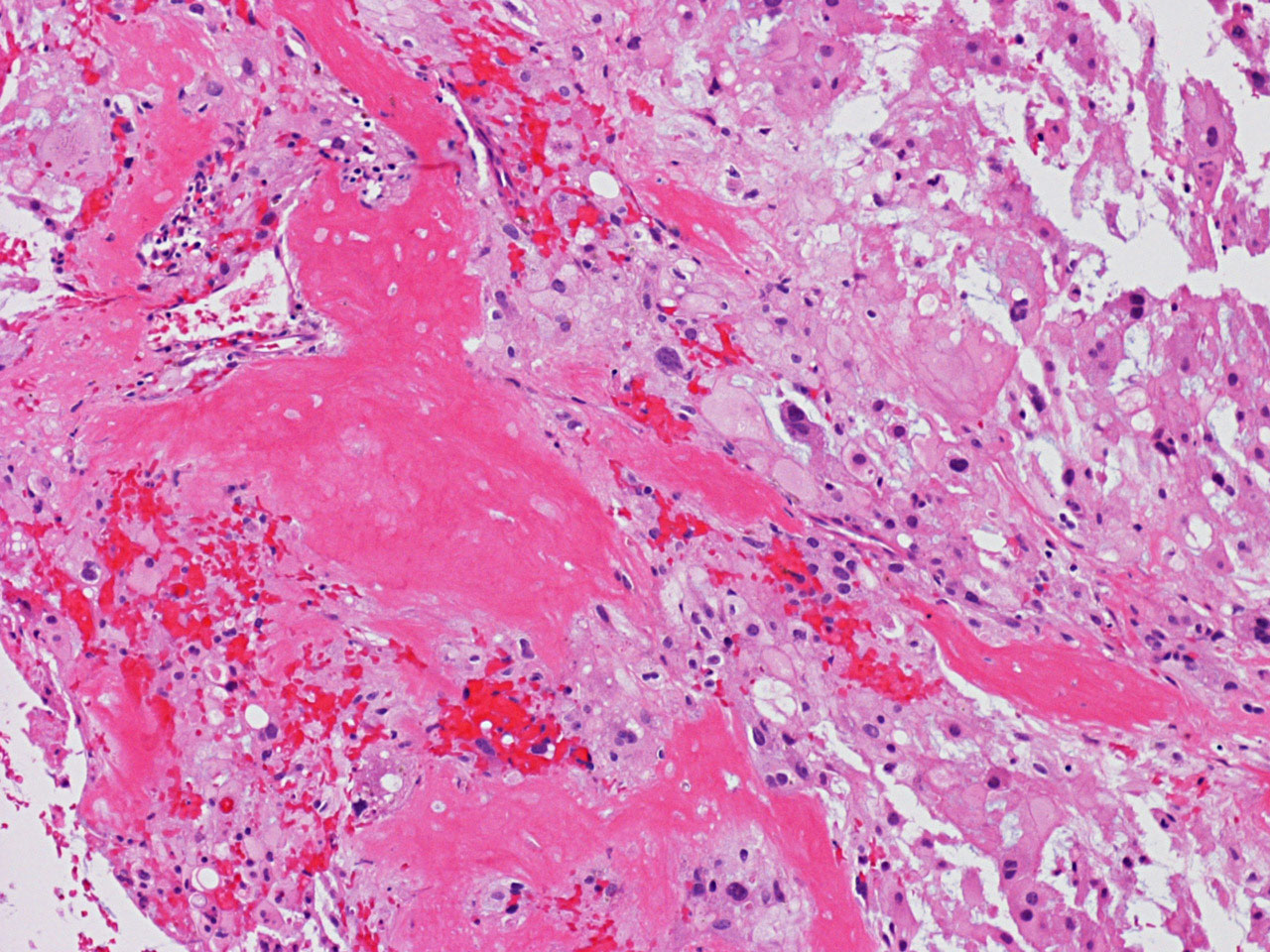
bubbles. The permanent sections (Panel G and H) show similar features. And the bubbly cytoplasm is more prominent
in the permanent sections (Panel H). Focal bone invasion is present (Panel I). The cytoplasmic vacuoles are best appreciated in the semithin
section (Panel M). Results of special studies are as follows:
Special Stain:
Immunohistochemistry:
Electron
microscopy:
| DIAGNOSIS: Chordoma |
Discussion: General
Information Pathology Immunohistochemistry Molecular
Pathology Differential
diagnosis Other
cases
Chordoma is an
uncommon malignant, slow growing, locally aggressive tumor with low metastatic
potential and histologic features of the notochord. In the World Health
Organization (WHO) classification, three distinct types are recognized namely
chordoma, chondroid chordoma, and dedifferentiated chordoma. All three are
malignant and has a high rate of recurrence. Transformation of chordoma into a high-grade spindle cell sarcoma
(dedifferentiation) has been described with and without radiation therapy
[Tsuboi Ys et al., 2007, Hanna SA et al., 2008].
Chordoma have
close histologic similarities with the notochord which is the purported origin
[McMaster ML, 2001]. However, it is unclear on whether all chordomas arise from
notochord remnants. The existence of soft tissue chordoma suggests that
notochord remnant is not a prerequisite for the development of chordoma [Lauer SR
et al, 2013 ]. Familial
clusters have been described
[Kelly MJ et al., 2001;
Wang et al.,
2015;
Kelly MJ et al., 2014]. Some are linked to chromosome 7q33
[Kelly MJ et al., 2001] and T (Brachyury) gene duplication
[Yang
XR et al., 2009;
Wang et al.,
2015]. Most cases are unifocal but rare multicentric cases have been
described
[Grossbach A et al., 2011, Lim JJ et al., 2009; Anderson WB & Meyers HI
1968].
After osteosarcoma, chondrosarcoma, and Ewing sarcomam, chordoma is the fourth most common osseous tumor. Similar to Ewing sarcoma, chordoma is less common in black patients. Sacrococcygeal and sphenooccipital regions are the most common location but it can be found along the entire axial skeleton. Within the skull, the clinoid region (clivus) is the most common. Occurrence in the sellar region as in this case is uncommon. Chordomas typically occur in the 5th and 6th decade but it can occur in all age groups include children. About 5% of the cases would occur before the age of twenty and they are typically skull base tumors [Hoch BL et al., 2006]. Most chordomas occur as osteolytic lesions and occasionally as sclerotic lesions on radiographic studies. Clinical manifestations typically rooted from its space occupying features.
Chordomas are locally aggressive tumors that tend to recur and have low metastatic potential. Metastases occur late in the disease course, with lungs and skin being the most common sites for disease spread. Chordomas have a poor response to conventional radiation therapy or chemotherapy, but are amenable to surgical excision. Survival is affected by the success or failure of local control. With its strategic cranial base location, complete surgical is often a challenge. Interestingly, chordomas are very response to proton therapy [Tauziede-Espariat A et al., 2016; Kabolizadeh P et al., 2017].
Grossly,
chordomas are soft to mucoid or gelatinous gray-tan masses. Its multilobulated
contour is better appreciated on imaging than on fragmented surgical specimens.
In contrast to chondrosarcoma arising in the skull base which tend to be
eccentrically locatred, chordomas typically arise along the midline.
The
characteristic histologic findings in chordomas are large polygonal cells with
distinct cell membrane and the vacuolated physaliphorous cytoplasm, the term
deriving from the Greek physalis, or "bubble. The vacuolated or
physaliphorous cells are best appreciated in cytologic smears or squash
preparations. Tumor cells grow in small nests and cords within a
myxoid/chondroid matrix and demonstrate round, sometimes rather uniform nuclei
with low nuclear-to-cytoplasmic ratios. The tumor cells tend to adhere into
clusters and cords. The classic large physaliphorous cell has a centrally
located nucleus surrounded by a narrow rim of cytoplasm that in turn, is
encircled by a ring of more peripherally located cytoplasmic vacuoles. Nuclear
grade is not particularly high in some cases but many of them have clearly
recognizable nuclear pleomorphism. Occasional large, atypical cells are present.
These nuclear changes should not be present in benign notochordal cell tumor and
ecchordosis physaliphora/fetal vestige
[Amer & Hameed, 2010].
On cytologic smears
[Crapanzano JP et al., 2001], chordoma cells tend to be cohesive but not
as cohesive to each other as carcinomas. Strings of chordomas are common
features.
Chondroid chordomas typically occur in the skull base. It contains matrix that
resemble hyaline cartilage and this component can be diffuse or focal
[Oakley GJ et al., 2008]. Dedifferentiation (dedifferentiated
chordoma) can occur and the dedifferentiated component appears as high-grade
sarcoma. Dedifferentiated chordoma usually compose of a well-demarcated
high-grade sarcomatous component arising in a background of chordoma. When no
residual low-grade component present, correct diagnosis can be a challenge. The
anatomical location and a high index of suspicion are good diagnostic help.
On
immunohistochemistry, nuclear expression of brachyury (a T-box transcription
factor encoded by the
TBXT
gene involved in notochordal development) is a highly specific marker when the
clinical and histopathologic features are taken into consideration
[Miettinen M et a., 2015; Oakley GJ et al., 2008; Vujovic S et al., 2006; Sangoi AR et al., 2011, Clayton EF et al., 2013]. With this said, one must note that nuclear expression of
brachyury is also expressed in about three quarter of the cases of embryonal
carcinoma, half of the cases of seminoma and a minor proportion of yolk sac
tumor, and 41% of small cell carcinoma
[Miettinen M et a., 2015]. Brachyury is also expressed in the cytoplasm of intracranial
hemangioblastoma
[Barresi V et al., 2012] but not in peripheral hemangioblastoma
[Doyle LA & Fletcher CD, 2014], primary carcinoma of lung
[Haro A et al., 2013], prostate cancer
[Pinto F et al., 2014], and colorectal carcinoma
[Kilic N et al., 2011]. Although brachyury is usually positive for chordoma,
immunoreactivity can be lost in decalcified tissues, and it is not typically
expressed in the dedifferentiated component of dedifferentiated chordomas.
Loss of
SMARCB1/INI1
(can be demonstrated by immunohistochemistry to BAF47) is common
[Buccoliero AM et al., 2019; Mobley BC et al., 2010; Antonelli M et al., 2017]
and is usually seen in poorly differentiated cases. In up to 50% of the
pediatric cases, INI1 is lost
[Antonelli M et al., 2017]. With the skull base location taken into consideration and the
high-grade histologic features, these tumors can be misdiagnosed as atypical
teratoid/rhabdoid tumor.
Chordomas are
typically positive for brachyury, pan-cytokeratin, epithelial membrane antigen,
SOX9, SHH, cathepsin K, and cadherin. Although the combination of characteristic
morphology with strong positivity for pan-cytokeratin are diagnostic for
chordoma until proved otherwise, one must note that chordomas are typically
negative for cytokeratin 7 and cytokeratin 20
[Folpe
AL et al., 1999]. Expression of cytokeratin 18 is
variable. Chordoma
is variably positive for S100. This is different from chondrosarcoma where S100
is typically evenly and strongly expressed. Ki67 labeling is moderate to high.
This is an important features to distinguish chordoma from benign notochordal
cell tumor and ecchordosis physaliphora/fetal vestige
[Amer and Hameed, 2010].
Genetics & Molecular Pathology:
Chordomas have
frequent cytogenetic abnormalities that include monosomy of chromosome 1 and
gain of chromosome 7. They show a near diploid to moderately hypodiploid
karyotype. Homozygous or heterozygous loss of CDKN2A and CDKN2B are
found in about 70% of cases
[Vujovic S et al., 2006]. Loss of PTEN and
amplification of EGFR are also seen
[Shalaby A et al., 2011]. Activating mutations in the mTOR pathway,
PDGFB, IGFR1 and
IGF1 are also identified. Somatic
mutations are very rare. One study found somatic mutations in
PIK3CA in 3 (of 287) low grade chordomas
[Tauziéde-Espariat
et al.,
2016].
Treatment:
Surgical
resection with a clear margin is still the best positive prognostic factor.
However, chordomas occurring in particularly skull base may make complete
resection impossible. If total resection is not possible, adjuvant radiation and
proton radiation may provide a good short-term outcome
[Kabolizadeh
P et al., 2016].
There are many ongoing clinical trials to explore potentially more treatment
options targeting the molecular pathways such as those involving in PD-L1
[https://clinicaltrials.gov/ct2/show/NCT03623854].
Prognosis:
Tauziéde-Espariat et al., (2016) proposed a histopathologic grading
system using histopathologic differentiation, mitotic count, apoptosis,
prominent nucleoli, necrosis, Ki67 count, and p53 expression as parameters. With
their scoring system, tumors are separated into low- and high-grade that reflect
prognosis.
Differential Diagnosis:
Benign
notochordal tumor vs. chordoma vs. ecchordosis physaliphora/fetal vestiges: Both benign
notochordal tumor and ecchordosis physaliphora [Amer
& Hameed, 2010] are typically under 4 cm and are often
incidental findings while chordomas are much larger and often symptomatic.
Radiographically, chordomas are more aggressive, usually with infiltration into
the surrounding soft tissue, while benign notochordal tumor and ecchordosis
physaliphora are non-invasive. Histologically, the basic histologic features are
similar but with subtle recognizable differences. Benign notochordal tumor
typically has fatlike clear cells, eosinophilicl cells, and physaliphorous
cells. Ecchordosis
physaliphora typically has strands of eosinophilic notochordal cells with small
pyknotic nculei in a myxoid background. Nuclear atypia is present in chordoma but not in the
other two entities. Chordoma also has classic lobular arrangement and fibrous
bands in between tumor cells. Myxoid background are present at least focally in
most cases. Mitotic fibures should only be seen in chordoma. Some chordomas can
be overtly pleomorphic in appearance with high grade nuclei. These cases should
not be a challenging situation. The immunohistochemistry profile is very similar except that
benign notochordal tumor are typically positive for cytokeratin 18 which is only
variably expressed in chordoma and ecchordosis physaliphora. The most important
difference is that chordoma has much higher Ki67 labeling index.
Parachordoma
(myoepithelioma/mixed tumor of soft tissue): First and foremost, parachordoma, must be distinguished from
genuine soft tissue chordoma which are positive for brachyury
[Lauer
SR et al., 2013]. Parachordoma is a rare tumor that has
not been reported to occur in the skull. They usually occur as subfascial mass
in thigh, arm, and chest wall. It is a tumor that has morphologic features of
both chondrosarcoma and chordoma. The peak incidence is the second to
fourth decades of life and a small number are noted in pediatric patients. Most
of them are seen in the head and neck region, often in a subcutaneous or deep
location. The histopathology is similar to myoepitheliomas occurring in salivary
glands. These tumors that are classically regarded as "parachordoma" demonstrate
a spectrum of morphology from spindle to epithelioid cells to pale staining
cells with collagenous to chondromyxoid stroma. Some of these contains small
nests of cells with pale to eosinophilic cytoplasm resembling chordoma cells and
grow in cords, chains, and clusters resembling myxoid chondrosarcoma. Some of
these cells may transform into spindle or round-globoid cells. The level of
nuclear atypia and mitotic count are both low. Immunohistochemically, these
tumors are negative for brachyury. They strongly express cytokeratins 8/18 but
not other cytokeratins. They also express EMA, S-100 protein, vimentin, CD34,
and type IV collagen
[Folpe
AL et al., 1999]. This profile, in fact, overlaps with
that of chordomas. Myoepithelial markers such as muscle specific actins, p63,
calponin, and glial fibrillary acidic protein (GFAP) are positive in some of the
cases
[Folpe
AL et al., 1999; Hornick
JL et al., 2003]. Last but not the least, many of these
tumors show rearrangement of the EWSR1 gene
[Flucke
U et al., 2012; Flucke
U et al. 2011; Antonescu
CR et al., 2010].
Metastatic
clear cell carcinoma and soft tissue chordoma: When chordoma is obtained from the classic locations, it is usually
not a major diagnostic challenge as the histopathology in most of these cases
are classic. The two major challenges include genuine soft tissue chordoma [Lauer
SR et al., 2013] and metastatic chordoma without
knowing the history. Chordomas are positive for cytokeratin. When this is
interpreted with the large round to polygonal epithelioid cells, soft tissue
chordoma and metastatic chordoma can be mistaken as metastatic clear cell
carcinoma. A high index of suspicion, informative clinical history, and
utilization of the appropriate immunohistochemical panel are the key to resolve
this situation.
Chondrosarcoma: Chondrosarcoma occurring in a location
where chordoma can be found may pose a diagnostic challenge, especially on small
biopsies. In the sacral and cranial base, chondrosarcomas are often eccentric
while chordoma is located almost perfectly along the midline. In contrast,
chordoma cells are large and have abundant eosinophilic cytoplasm. Their
numerous desmosome-type intercellular junctions cause the cells to be tightly
opposed to one another without intervening matrix, producing cohesive nests or
sheet-like structures. In squash preparations, they may appear as short strings
of cells. Chordoma cells also like to wrap around another chordoma cells as if
one is "hugging" the other. It is not uncommon for the tumor cells like
chondrosarcoma and choroid glioma to have to have bubbly cytoplasm. Genuine
lacunae are not seen in chordoma but in chondrosarcoma. This is important
because most chondrosarcoma arising from the cranial base are low-grade tumor
with well-formed lacunae. Chondrosarcomas are typically immunoreactive for D2-40
(podoplanin) and EMA, but negative for pancytokeratin and glial fibrillary
acidic protein (GFAP). In contrast, chordoma is typically positive for
brachyury, EMA and pan-cytokeratin, but negative for D2-40 and GFAP. About 50%
of chondrosarcomas harbors mutation of IDH1 and IDH2
[Aria
M et al., 2012], which is not seen in
chordoma Podoplanin is positive for chondrosarcoma but not chordoma
[Oakley
GJ et al., 2008].
Atypical
teratoid/rhabdoid tumor (AT/RT): Lost of INI1 on immunohistochemistry is common and is present in
about half of the cases of pediatric chordomas [Buccoliero
AM et al., 2019; Mobley
BC et al., 2010; Antonelli
M et al., 2017]. This occurs often in poorly differentiated cases.
This may post a diagnostic pitfall as AT/RT is
characterized by loss of INI1. AT/RT is usually seen in infants and less
commonly in older children. Chordoma is usually seen in adult. AT/RT is usually
a tumor within the parenchyma (intr-axial) of the brain. Chordoma is often
intraosseous or has an intraosseoous component. Histologically, they are very
different. AT/RT is polyphenotypic and is positive for synaptophysin, glial
fibrillary acidic protein, neurofilament and other markers, while negative for
brachyury.
Dedifferentiated chordoma: The presence
of components of a typical chordoma or a prior history of chordoma with
recurrence are both helpful in making this diagnosis. The dedifferentiated
chordoma would have histology of a high-grade spindle cell sarcoma and the
characteristic immunohistochemical profile of a chordoma may not be detected,
such as brachyury may be lost in the dedifferentiated component It should be
noted that de novo dedifferentiated chordomas have been described
[Hanna
SA, 2008].
Chordoid
meningioma: Meningioma arising at the base of
skull may invade into bone. In these cases, the bulk of the tumor is mostly
extra-osseous and intra-cranial. Pure intra-osseous meningioma can occur but is
rare
[Abuzayed
B et al., 2019; Butscheidt
S et al, 2019]. Chordoid meningioma is characterized
by areas with stroma vaguely reminiscent of chordoma and within this background
are neoplastic epithelioid to round, polygonal meningothelial cells with pale
cytoplasm vaguely reminiscent that of chordoma. The matrix is positive for
Alcian blue. When this component exceeds 50% of the volume of the tumor, a
chondroid meningioma of WHO grade II can be made. It is rare to see pure
chordoid meningioma that would suggest chordoma. Most of them are admixed with
classic meningothelial meningioma or other patterns. Chordoid meningiomas are
positive for somatostatin receptor 2a (SSTR2a), EMA, progesterone receptor and
negative for brachyury.
Chordoid
glioma: This tumor occurs in the third
ventricle and the chordoid matrix vaguely suggest a chordoma, but the location
is a big help. In addition, these tumors are positive for glial fibrillary
acidic protein and negative for brachyury.
Myxopapillary
ependymoma: Myxopapillary
ependymomas occur most commonly in the cauda equina but not as intra-osseous
tumor. It is a WHO grade I tumor and does not invade bone. Other than the mucoid
component that may suggest chordoma, it has very little histopathologic
resemblance with chordoma. Myxopapillary ependymoma is negative for brachyury
and positive for glial fibrillary acidic protein.
Other Cases: