T1 + Contrast
FLAIR

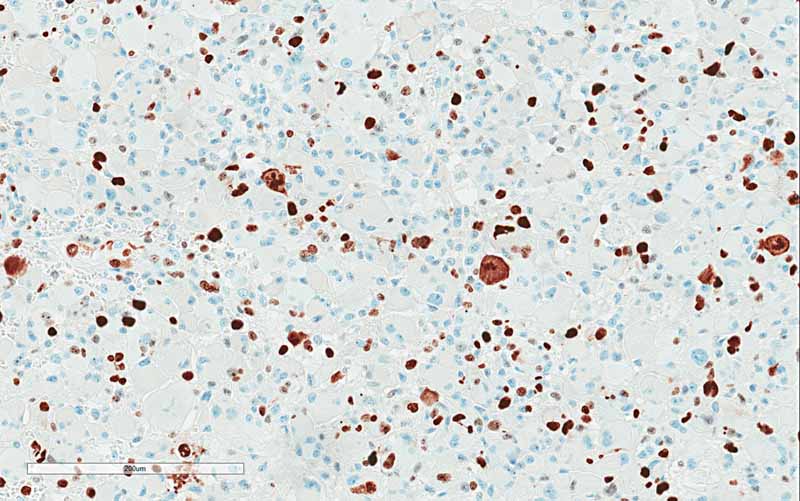
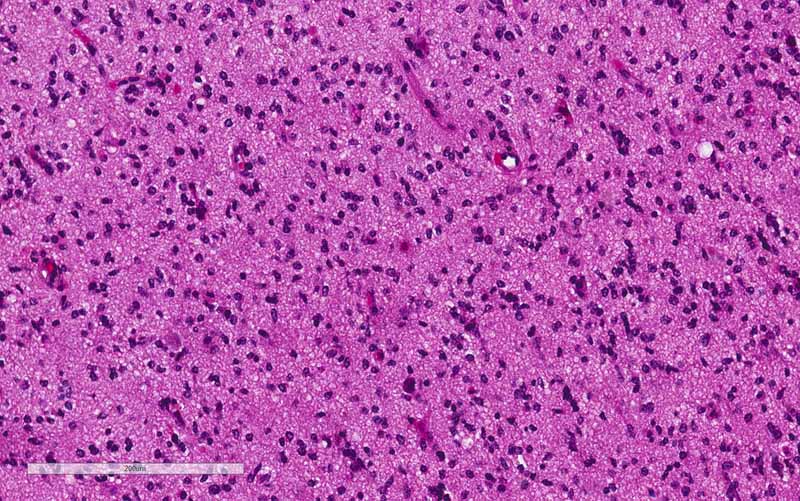
Ki67
Ki67
| A 76 year-old man with a History of Parkinson
Disease, Sudden onset of Weakness with Tremor. January, 2020, Case 2001-1. Home Page |
Kar-Ming Fung, M.D., Ph.D. Last update: April 30, 2020.
Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma.
Clinical information: The patient was a 76 year-old man with a history of Parkinson's disease. He developed sudden onset weakness of lower extremities while standing. He did not fall and was able to sit down. There was a brief tremor of his upper extremities but there was no seizure. On furhter work up, an enhancing mass, 2.0 x 1.6 cm, was noted in his right temporal pole. The mass was resected.
|
|
|
|
|
|
|
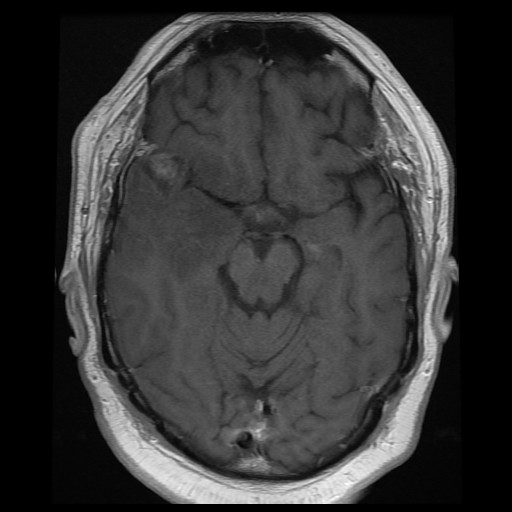
A. T1 + Contrast |
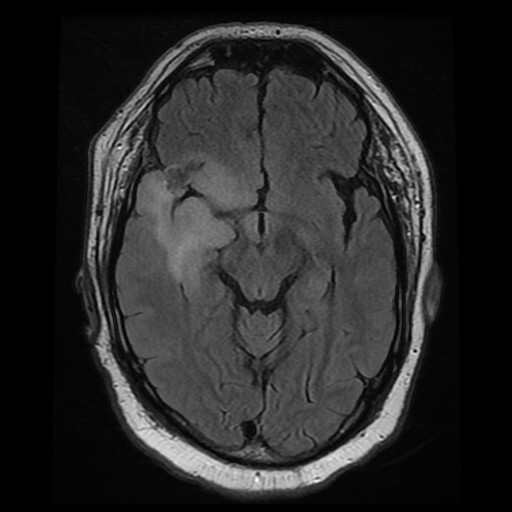
B. FLAIR |
C. | D. | E. |
|
|
|
|
|
|
| F. | G. |
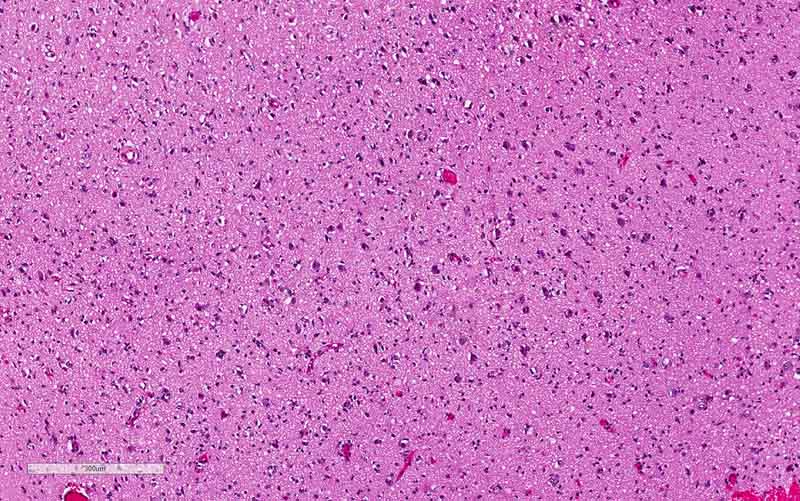
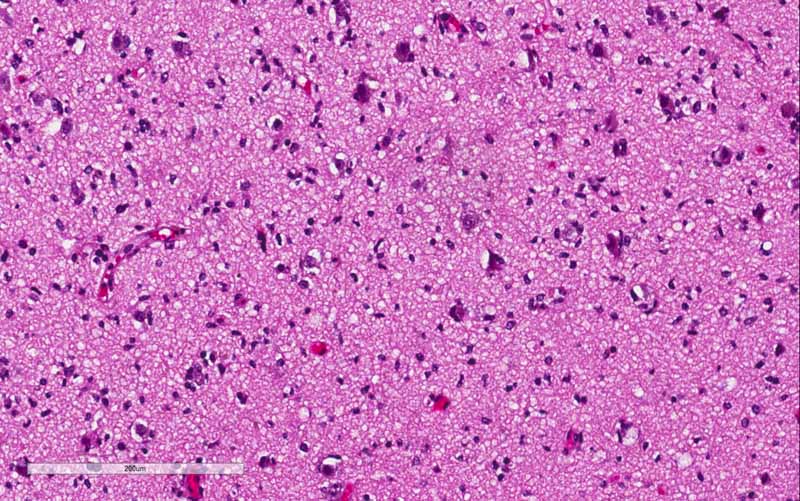
H. Ki67 |
I. Ki67 |
Radiology
of the Case: The mass is
located at the tip of the right temporal lobe causing minimal midline
shift. There is heterogensous enhancement suggestive of necrosis
(Panel
A).
There is substantial edema around this mass
with extension inot the posterior temporal lobe, insula, and inferior
lateral frontal lobe
Comment on
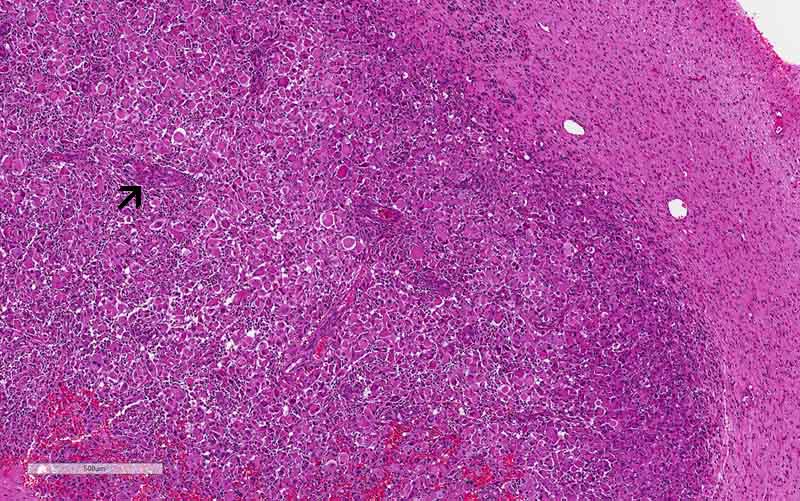
pathology: The rhabdoid area is
well-demarcated fromt he surrounding high-grade glioma with a pushing margin.
This phenomenon is most consistent with a high-grade/rhabdoid transfromation
arising from the surrounding high-grade glioma areas. This kind of
well-demarcated high-grade changes is more commonly seen in malignant/high-grade
transformation in sarcomas.
Molecular profile: A paraffin block containing the rhabdoid changes was
submitted for next generation sequencing and chromosome microarray testing.
Mutations:
TERT,
EGFR,
KRAS,
KDM61
Chromosomal abberations: +7, EGFR
amplification, CDKN2A/B homozygous deletion, and -10
MGMT promoter methylation: None
| DIAGNOSIS: Glioblastoma, WHO grade IV, IDH wild-type, with rhabdoid changes |
Discussion:
General Information
Pathology Rhabdoid
Changes Differential diagnosis
Related Interesting Cases
The prefix
rhabdo- is borrowed from Greek and it
means rod-like, rods, or wands. Perhaps it is a good way to describe the
striations in skeletal muscle, it is used to describe skeletal muscle type of
differentiation. Before the introduction of immunohistochemistry and molecular
biology, pathologists have to hunt for the striations the emulate striations of
skeletal muscle in a tumor in order to call it a rhabdomyosarcoma [click here to see a case]
or rhabdomyoma [click here to see a case].
In the modern practice, immunohistochemistry is typically employed to
demonstrate that there is no expression of desmin, myogenin, and MyoD for a cell
or tumor to be called rhabdoid.
Rhabdoid, on the other
hand, is a term used to describe cells that have features mimicking skeletal
differentiation but they are not they are not real skeletal differentiation.
Desmin, myogenin, MyoD and other skeletal muscle markers cannot be demonstrated
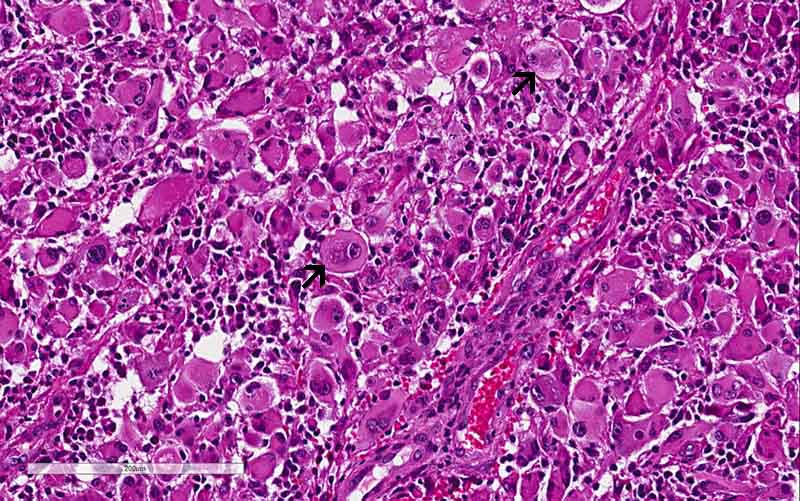
in these tumors. Rhabdoid cells are large polygonal cells with a large central
inclusion ultrastructrally composed of intermediate filament that is usually
vimentin. Nuclei are large, vesicular, with prominent nucleoli, and are
eccentric to the inclusion.
Primary rhabdoid tumors
constitute a group of predominantly pediatric tumors that have features
morphologically suspicious of skeletal muscle but without genuine skeletal
muscle phenotype. These tumors are mostly seen in kidney, in soft tissue and in
the central nervous system (atypical teratoid/rhabdoid) tumor. However they have
been described in virtually all of the anatomical sites. Most of these tumors
share the genetic signature of deletion of
SMARCB1 gene
(also known as hSNF5/INI1/BAF47)
on chromosome 22 with a small number of atypical teratoid/rhabdoid tumors
associated with loss of BRG1 (SMARCA4) rather than
INI1. The protein product can be detected by immunohistochemistry
[Judkins AR, 2007]. Histologically,
epithelioid tumor cells have certain similarities with rhabdoid tumor cells.
Many epithelioid tumors are associated with loss of
INI1 expression [Hornick
JL et al., 2009].
Rhabdoid change, however, can be seen as a subpopulation of cells arising from a
histologically lower grade tumor [Perry
A et al., 2005,
Fung et al., 2004]. It is an indication of
malignant or high-grade transformation. It is essentially a growth pattern but
not a differentiation. It is typically not associated with loss of INI1
expression. Clinically, rhabdoid changes are typically associated with rapid
enlargement of a pre-existing tumor [Fung
KM et al., 2004]
and rapid clinical decline. These tumor are referred to composite rhabdoid
tumors and rhabdoid changes can occur in a wide variety of tumors.
Characteristically, no loss of INI1 is present in these composite rhabdoid
tumors [Perry A et al., 2005].
There a rare report of a glioblastoma with rhabdoid changes associated with loss
of INI1 in the rhabdoid component [Yamoto
J et al., 2011].
Rhabdoid glioblastoma
refers to tumor with rhabdoid cells arising within a background of otherwise
classic glioblastoma. It fits the definition of composite extra-renal rhabdoid
tumor. It should not be confused with epithelioid glioblastoma which is composed
of solid sheets of discohesive epithelioid cells without association with a
conventional glioblastoma component. Phenotypically, both rhabdoid and
epithelioid glioblastoma have intact INI1 and they are non-reactive for claudin
6 which is a marker for atypical teratoid/rhabdoid tumor
[Kleinschmidt-DeMasters
BK et al., 2010].
Both rhabdoid and epithelioid glioblastoma are more aggressive than conventional
glioblastoma [Sugimoto K et al., 2016].
Next generation sequencing panel study and chromosome microarray study were
performed in this case with a block containing the rhabdoid changes. There was
no loss of SMARCB1(INI1)
or SMAECA4(BRG1) were
demonstrated by next generation sequencing. Chromosomal changes included
gain of chromosome 7,
EGFR amplification,
CDKN2A/B homozygous deletion, and
heterozygous loss of chromosome 10. There was
also mutation of TERT promoter. These
features were classic for a glioblastoma. The rhabdoid changes stood out
distinctly in a background of high-grade glioma. The significant increase
in Ki67 labeling in the rhabdoid area. Both features supported that the rhabdoid
component was a high-grade transformation arising from the existing glioma. With
all of these features taken into consideration, this is a case of glioblastoma
with rhabdoid changes.
Glioblastoma with Rhabdoid Changes:
Glioblastoma, formerly
known as glioblastoma multiforme, has a reputation to demonstrate a wide range
of histopathologic patterns. In the 2016 World Health Organization (WHO)
classification of brain tumors, glioblastomas are separated into two major
groups, the IDH wild-type and
IDH mutant. In tumors with
IDH wild-type, there are several
patterns in addition to the classic pattern of high-grade glioma with
endothelial proliferation and, often, pseudopalisading necrosis. These patterns
include small cells and small cell glioblastoma, primitive neuronal cells and
glioblastoma with a primitive neuronal component, glioblastoma with
oligodendroglioma component, glioblastoma with gemistocystic astrocytic
components, glioblastoma with multinucleated giant cells, granular cells and
granular cell glioblastoma, lipidized clls and heavily lipidized glioblastoma,
and metaplastic glioblastoma.
In the 2016 WHO
classification, WHO also recognizes three rare variants namely epithelioid
glioblastoma, giant cell glioblastoma, and gliosarcoma. These variants typically
lack IDH mutation. Epithelioid glioblastoma is a characterized by mitotically
active, closely packed epithelioid cells and some rhabdoid cells with
microvascular proliferation and necrosis. Giant cell glioblastoma is
characterized by bizarre, multinucleated giant tumor cells and an occasionally
abundant reticulin network. Gliosarcoma is characterized by a biphasic pattern
with components demonstrating glial and mesenchymal differentiation.
Neither glioblastoma
with rhabdoid changes nor rhabdoid glioblastomas are recognized as a specific
pattern or variant in the 2016 WHO classification. Therefore, glioblastoma with
rhabdoid change (or component) is a better term than rhabdoid glioblastoma for
the current tumor which has both a conventional high grade glioma component and
a distinct subpopulation of rhabdoid tumor.
Yamamoto J et al.
described a similar case in 2012. Like other composite rhabdoid tumors, the
rhaboid component is signature of rapid growth. INI1 genes, like the current
case, is typically preserved [Perry A et al.,
2005,
Fuller CE et al.,
2001] but there are
exceptions [Donner LR et al., 2007,
Wyatt-Ashmead J et al., 2001].
Differential Diagnosis:
Epithelioid
glioblastoma: As mentioned earlier, epithelioid glioblastoma and
rhabdoid glioblastoma share some cytologic similarities. One can also imagine
that rhabdoid change is an exagerated version of epithelioid changes. However,
epithelioid glioblastoma is composed entirely of epithelioid cells while
rhabdoid glioblastoma contains rhabdoid areas embedded within a background of
conventional glioblastoma.
Metastatic rhabdoid carcinoma: In the personal experience of the
author, metastatic carcinoma with rhabdoid changes are rare biopsy specimens
fromt he brain. It is true that rhabdoid tumors are more aggressive and they
probably are more likely to metastasize. The speculation is that they would kill
the patient so fast that biopsy or resection of tumor in the brain are not
performed for many reasons. Anyway, metastatic rhabdoid carcinoma, however,
looks homogeneously rhabdoid which may be more suggestive of epithelioid
glioblastoma than rhabdoid glioblastoma. In addition, they can be multiple and
may have a positive history. Besides, they should be negative for glial
fibrillay acidic protein wile rhabdoid and epithelioid glioblastoma are
typically positive [Sugimoto K et al., 2016].
Atypical
teratoid/rhabdoid tumor [click
here to see a classic case]: This tumor occurs mostly in infants
under 3 years of age but uncommon in older children, and rare in young adults.
In contrast to its name, the degree of rhabdoid changes can vary from minimal to
fully developed. Most of them have loss of INI1 which can be demonsrated on
immunohistochemistry. But this is not full proof as a small number of them have
loss of BRG1. In coa ntrast to rhabdoid
glioblastoma, rhabdoid cells in atypical teratoid/rhabdoid tumors do not form a
distinct area that stands out from the rest of the tumor. One must also note
that they can be positive for GFAP and which can be confused with rhabdoid or
epithelioid glioblastoma.
Metastatic
melanoma: Metastaic melanoma to the brain can appears as a
discohesive plasmacytoid or epithelioid tumor. These
tumors can, in fact, looks like a plasma cell tumor on cytologic preparation but
the nuclear grade is usually higher than that of a melanoma. Many of them are
amelanotic. However, metastatic melanoma will not be associated with a
conventional glioblastoma. On immunohistochemistry, melanoma are positive for
MART-1 (MelanA), tyrosinase, HMB45, and MiTF. These markers are
negative for glioma.
Related
Interesting Cases: