EGFR FISH
| A 55 year-old Man with
a Parietal Enhancing Mass. November, 2003, Case 311-1. Home Page |
Gregory Blakey, M.D.1, Arie Perry, M.D.2, Kar-Ming Fung, M.D., Ph.D. 1, Last update: March 30, 2004.
1 Department of Pathology, University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma, and 2 Department of Pathology, Washington University, St. Louis, Missouri
Clinical information: The patient was a 55 year-old man who presented with headache. A 3 cm parietal intraparenchymal mass with rim enhancement was noted on MRI scan. A stereotactic biopsy was performed.
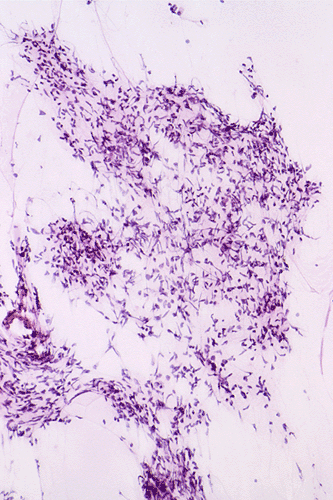
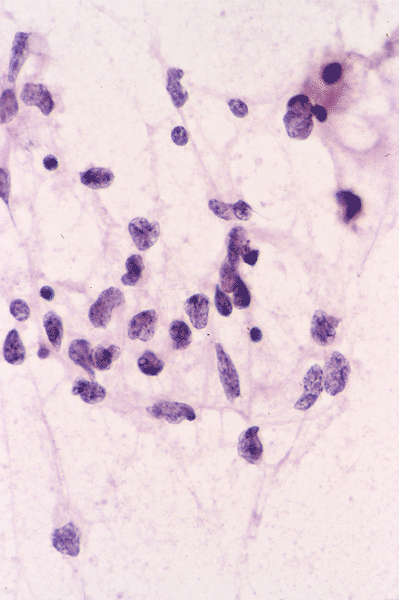
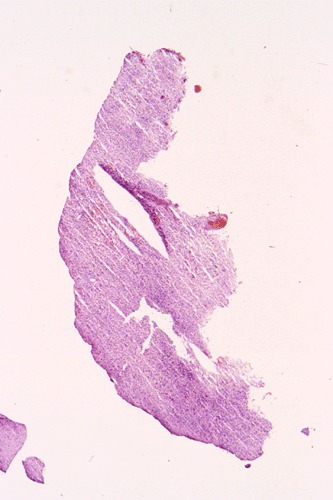
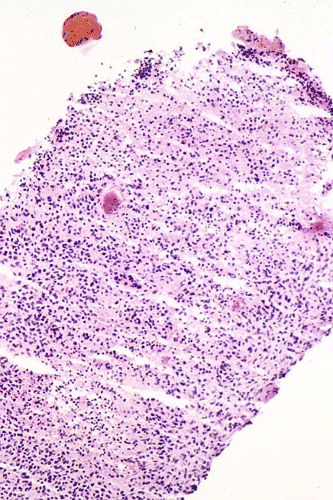
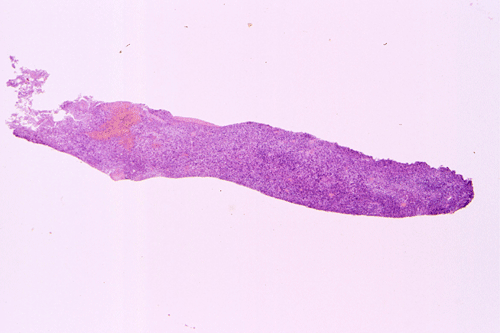
The following photos are taken from representative areas. Panel A and B are taken from intro-operative cytologic preparation. Panel C to F are taken from the frozen section. Panel G to I are taken from formalin fixed paraffin embedded material that has not been frozen. Panel J is fluorescent in situ hybridization (FISH) studies on epidermal growth factor receptor (EGFR).
|
|
|
|
|
|
|
| A. | B. | C. | D. | E. | F. |
|
|
|
|
|
||
| G. | H. | I. |
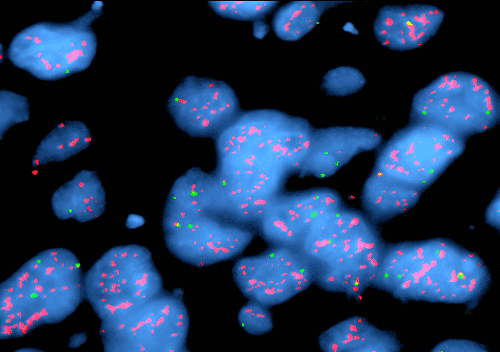
J. EGFR FISH |
Pathology of the case:
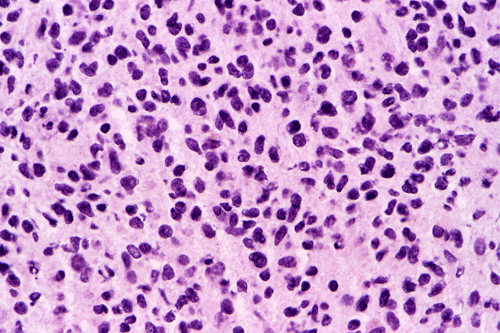
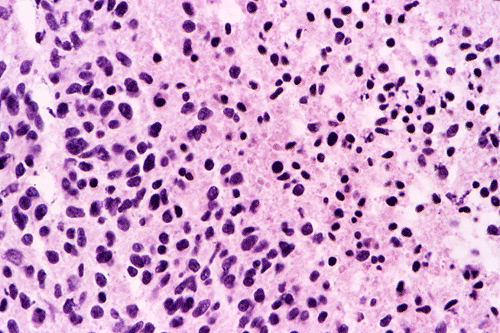
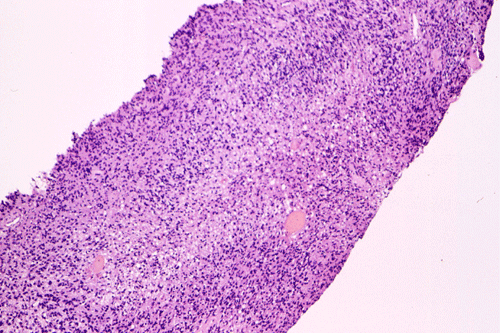
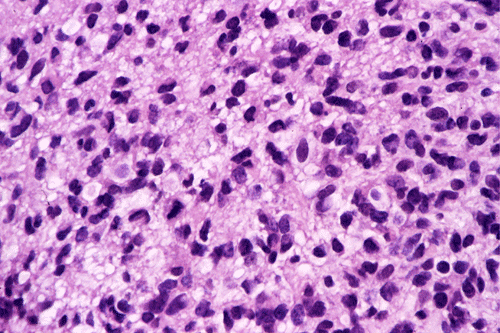
Intraoperative consultation: The cytologic preparation provides some very useful information. The cells form poorly defined clumps with elongated, pale pink cytoplasmic processes (Panel A, arrow). The nuclei are elongated, hyperchromatic and resemble "baking potato". Nucleoli are not seen (Panel B), On the frozen section, a small streak of pale pink tissue suggestive of necrosis is present (Panel C and D). On high-magnification, the viable tumor cells have medium to large amount of cytoplasm and without distinct cytoplasmic border. They are densely packed, with hyperchromatic nuclei and does not form a specific pattern (Panel E). The interphase between the necrotic cells and the viable cells on frozen section is illustrated in (Panel F). The intraoperative diagnosis was "high-grade glial neoplasm with necrosis, probably glioblastoma".
Permenant section: The pathologic changes of the permenant sections are similar to that of frozen. Again, pseudopalisading necrosis is present (Panel G and H). Many of the nuclei are elongated and there is also considerable degree of pleomorphism. Some of the cells have perinuclear clearing (Panel I). Positive staining for glial fibrillary acidic protein (GFAP) but not synaptophysin is demonstrated by immunohistochemistry. Although the perinuclear halo may suggestive anaplastic oligodendroglioma, the overall pathologic features, particularly the elongated nuclei and degree of pleomorphism, are that of a glioblastoma.
FISH: There was amplification of epidermal growth factor receptor (EGFR) (Panel J) and loss of chromosome 10q. No deletion of chromosome 1p and 19q was found. These findings are more suggestive of an glioblastoma rather than anaplastic oligodendroglioma.
|
DIAGNOSIS: Glioblastoma (WHO grade IV/IV) . |
Discussion: General Information Imaging Pathology Molecular pathology Differential diagnosis
General Information
Glioblastoma is one of the most malignant tumors among known human neoplasms and unfortunately one of the most common malignant brain tumors. Glioblastoma represents about 12-15% of all intrancranial tumor and about half of the number of cerebral glial tumors 1. The peak age of occurrence is typically later than that of diffuse astrocytoma and anaplastic astrocytoma with a peak incidence in the 6th and 7th decades. It can occur also in other ages and about 10% of the tumors occur in children and a small number of them in infants. An increased incidence of glioblastoma is observed in Turcot syndrome 2. The prognosis of glioblastoma is grave and 2-year survival rate is less than 10% even with therapy. Massive local recurrence is common.
MRI imaging usually reveals a poorly delineated mass with low T1 signal intensity. “Ring enhancement” formed by a rim of enhancing and viable tumor with a non-enhancing, often necrotic, core is the most common finding. Significant vasogenic edema is often demonstrated by T2-weighed and FLAIR images. The margin of the tumor is often difficult to be determined due to the infiltrative nature and edema of these tumors. Hemorrhage or blood products are common. Mass effect leading to midline shift and herniation is common. Glioblastomas often extend across the midline through the corpus callosum and involve the other cerebral hemisphere, the so-called “butterfly tumor”.
Although glioblastoma is a very infiltrative tumor, the hemorrhage and necrotic components may lead to a relatively circumscribed macroscopic appearance. Infiltrating tumor without associated necrosis and hemorrhage will blur or obliterate the anatomical landmarks. Extensive edema can be resulted from a relatively small tumor. Therefore, macroscopic determination of the extent of the tumor is often difficult and inaccurate. Thrombosed blood vessel is a well-recognized intraoperative observation under the dissecting microscope and also at the dissection bench. Particularly, substantially thickened blood vessels with thrombus are common in or near the necrotic center. Infiltration across the midline through the corpus callosum will lead to expansion, sometimes hemorrhage and necrosis, of the corpus callosum. Infiltrating of the cortex, adjacent leptomeninges and dura are more common in the superficially located tumors.
Glioblastoma has been known as glioblastoma multiforme for many years because of its diversity in histologic appearance.. The tumor cells can vary from relatively small cells to large, pleomorphic cells to multinucleated giant cells. There is significant variation between different histologic fields and different tumors. In general, most tumor cells retain some morphologic features of glial cells particularly astrocytes. Endothelial proliferation is found in most case. Necrosis can be extensive. Pseudopalisading necrosis is a common finding and refers to necrosis, often slit like and Y-shaped, that is surrounded by fusiform tumor cells arranging in a palisading fashion. Mitotic activity varies from moderate to brisk and atypical mitoses, more commonly found in tumor with large and anaplastic cells, can be present. As per the diagnostic criteria of the World Health Organization (WHO), the presence of anaplasia, increased mitotic activity, and endothelial proliferation with or without necrosis are required for the diagnosis of glioblastoma 1.
Immunohistochemically, glial fibrillary acidic protein (GFAP) is often detectable at least focally in most cases. However, the level of expression is inversely related to the degree of anaplasia. Focal expression of cytokeratin is also present which makes cytokerain alone a relatively non-specific marker to differentiate glioblastoma from poorly differentiated metastatic carcinoma. Immunohistochemistry for Ki67 (MIB1 antibody) will reveal a labeling index that varies from 5% to over 20%.
Like many human tumors, the pathogenesis of gliomas involves accumulation of multiple steps of genetic aberrations leading to inactivation of tumor suppressor genes and activation of oncogenes. Two receptors that utilize tyrosine kinase signaling pathway, namely platelet-derived growth factor receptor alpha (PDGFR-a) and epidermal growth factor receptor (EGFR), are closely related to gliomas pathogenesis. PDGFR-a is located at chromosome 4q11-12 and is over expressed in low- and high-grades of gliomas. Overexpression of PDGFR-a is considered an early event in gliomas pathogenesis. Genetic amplification of EGFR is a feature in about half of the all the cases of glioblastoma and in some anaplastic astrocytomas. Particularly, it is most common in primary (de novo) glioblastomas that usually arise in older adults without a pre-existing low-grade astrocytoma. Genetically, primary glioblastomas are less associated with p53 mutations. Interestingly, EGFR amplification is also associated with small tumor cells as illustrated in our case.
Secondary glioblastoma refers to glioblastomas that arise in the setting of pre-existing low-grade gliomas, most often an astrocytoma. They are more common in younger patients. The common genetic aberrations include mutation of p53 (LOH 17q), p16 (LOH 9p), Rb (LOH 13q), and LOH 19q 3, 4.
Loss of chromosome 10 is common in both primary and secondary glioblastomas and other high-grade gliomas. Mutation of PTEN, located at chromosome 10q23.3 is found in about 30% of primary glioblastoma and 5% in secondary glioblastoma 5, 6. Interestingly, loss of heterozygosity (LOH) at the PTEN locus is found in 75% to 90% of glioblastomas 7. Such discrepancy between LOH of chromosome 10 and mutation frequency of PTEN suggest that other genes in the same region, such as MXI1 and DMBT1 gene, are involved 7, 8. Other common deletions in high-grade astrocytomas include chromosome 19q 9 and 22q 10.
Deletion of chromosome 1p and 19q is present in up to 90% of oligodendrogliomas (WHO grade II) and 50-70% of anaplastic oligodendroglioma (WHO grade III). Oligodendrogliomas with deletion of chromosome 1p/19q tend to have the more “classic” appearance of oligodendroglioma, more responsive to chemotherapy, and have better prognosis. In cases with ambiguous histopathology, demonstration of 1p/19q deletion is a helpful mean in confirmation of oligodendroglial differentiation.
Differential diagnosis
The discussion on differential diagnoses will be restricted primarily to pathologic process that could be confused with the case under discussion. The biopsy material clearly revealed a neoplastic process that eliminates other non-neoplastic processes such as evolving infarcts and demyelinating diseases as possibilities.
In the current lesion, the pathology is quite classic for a malignant neuoepithelial tumor. The small cells, however, may suggest a diagnosis of primitive neuroectodermal tumor (PNET). The age of the patient is unusual for a supratentorial PNET. Although pseudopalisading necrosis can be seen in PNETs and medulloblastoma, they are relatively uncommon. PNETs and medulloblastomas are usually positive for synaptophysin by immunohistochemistry and also possess a large number of mitotic figures and apoptotic figures. Diffuse positive staining on immunohistochemistry is quite uncommon for PNETs.
The small tumor cells and occasional perinuclear halo are also suggestive of oligodendroglial tumor. Although pseudopalisading necrosis can be occasionally seen in anaplastic oligodendrogliomas, they are relative uncommon in comparison with glioblastoma. The nuclei in this case are not quite as round as that usually occur in oligodendroglioma and the increased pleomorphism is also more compatible with glioblastoma. Demonstration of EGFR amplication and loss of chromosome 10q without deletion of chromosome 1p and 19q are highly consistent with glioblastoma.
Metastatic small cell carcinoma is a concern in this age range. Morphologically, the neoplastic cells of this case lack the epithelioid/epithelial appearance of metastatic small cell carcinoma. Metastatic small cell carcinoma are usually positive for synaptophysin and negative for GFAP on immunohistochemistry. Other markers for epithelial cells such as epithelial membrane antigen (EMA) and cytokeratins may be used for differential diagnosis. It should be noted both reactive and neoplastic astrocytes can be strongly positive for cytokeratin, particularly AE1/AE3 as demonstrated by immunohistochemistry. In contrast, very few glioblastomas express low-molecular weight cytokeratin such as Cam 5.2. Glioblastomas are usually negative for EMA. The interpretation of immunohistochemical results should be cautious.
Reference:
Kleihues P et al., Glioblastoma. In Kleihues P and Cavenee, World Health Organization Classification of Tumours: Pathology and genetics, tumors of the nervous system. IARC press, Lyon 2000, page 29-39.
Paraf F, Jothy S, Van Meir EG. Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol. 1997;15:2744-58.
Bruner JM. Molecular markers in diagnosis and classification. In Zhang W, Fuller GN, Genomic and molecular neuro-oncology. Pg.81-93. Jones and Bartlett Publishers, Boston, 2004.
Davies M. PTEN, a tumor suppressor gene frequently mutated in glioblastomas. In Zhang W, Fuller GN, Genomic and molecular neuro-oncology. Pg.47-75. Jones and Bartlett Publishers, Boston, 2004.
Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol. 1998; 57:684-9.
Fujisawa H, Reis RM, Nakamura M, Colella S, Yonekawa Y, Kleihues P, Ohgaki H. Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab Invest. 2000; 80:65-72.
Fults D, Pedone CA, Thompson GE, Uchiyama CM, Gumpper KL, Iliev D, Vinson VL, Tavtigian SV, Perry WL 3rd. Microsatellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int J Oncol. 1998;12:905-10.
Mollenhauer J, Wiemann S, Scheurlen W, Korn B, Hayashi Y, Wilgenbus KK, von Deimling A, Poustka A. DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat Genet. 1997; 17:32-9.
Smith JS, Tachibana I, Lee HK, Qian J, Pohl U, Mohrenweiser HW, Borell TJ, Hosek SM, Soderberg CL, von Deimling A, Perry A, Scheithauer BW, Louis DN, Jenkins RB. Mapping of the chromosome 19 q-arm glioma tumor suppressor gene using fluorescence in situ hybridization and novel microsatellite markers. Genes Chromosomes Cancer. 2000; 29:16-25.
James CD, Carlbom E, Dumanski JP, Hansen M, Nordenskjold M, Collins VP, Cavenee WK. Clonal genomic alterations in glioma malignancy stages. Cancer Res. 1988; 48:5546-51.
Cases of the Month Evaluation Coordinator: KarMing-Fung@ouhsc.edu